Structural DNA helical parameters from MD trajectory tutorial using BioExcel Building Blocks (biobb)
Based on the NAFlex server and in particular in its Nucleic Acids Analysis section.
This tutorial aims to illustrate the process of extracting structural and dynamical properties from a DNA MD trajectory helical parameters, step by step, using the BioExcel Building Blocks library (biobb). The particular example used is the Drew Dickerson Dodecamer sequence -CGCGAATTCGCG- (PDB code 1BNA, https://doi.org/10.2210/pdb1BNA/pdb). The trajectory used is a 500ns-long MD simulation taken from the BigNASim database (NAFlex_DDD_II entry).
Settings
Biobb modules used
biobb_dna: Tools to analyse DNA structures and MD trajectories.
Auxiliary libraries used
jupyter: Free software, open standards, and web services for interactive computing across all programming languages.
matplotlib: Comprehensive library for creating static, animated, and interactive visualizations in Python.
Conda Installation & Launch
git clone https://github.com/bioexcel/biobb_wf_dna_helparms.git
cd biobb_wf_dna_helparms
conda env create -f conda_env/environment.yml
conda activate biobb_wf_dna_helparms
jupyter-notebook biobb_wf_dna_helparms/notebooks/biobb_wf_dna_helparms.ipynb
Pipeline steps
![]()
Initializing colab
The two cells below are used only in case this notebook is executed via Google Colab. Take into account that, for running conda on Google Colab, the condacolab library must be installed. As explained here, the installation requires a kernel restart, so when running this notebook in Google Colab, don’t run all cells until this installation is properly finished and the kernel has restarted.
# Only executed when using google colab
import sys
if 'google.colab' in sys.modules:
import subprocess
from pathlib import Path
try:
subprocess.run(["conda", "-V"], check=True)
except FileNotFoundError:
subprocess.run([sys.executable, "-m", "pip", "install", "condacolab"], check=True)
import condacolab
condacolab.install()
# Clone repository
repo_URL = "https://github.com/bioexcel/biobb_wf_dna_helparms.git"
repo_name = Path(repo_URL).name.split('.')[0]
if not Path(repo_name).exists():
subprocess.run(["mamba", "install", "-y", "git"], check=True)
subprocess.run(["git", "clone", repo_URL], check=True)
print("⏬ Repository properly cloned.")
# Install environment
print("⏳ Creating environment...")
env_file_path = f"{repo_name}/conda_env/environment.yml"
subprocess.run(["mamba", "env", "update", "-n", "base", "-f", env_file_path], check=True)
print("👍 Conda environment successfully created and updated.")
# Enable widgets for colab
if 'google.colab' in sys.modules:
from google.colab import output
output.enable_custom_widget_manager()
# Change working dir
import os
os.chdir("biobb_wf_dna_helparms/biobb_wf_dna_helparms/notebooks/abc_setup")
print(f"📂 New working directory: {os.getcwd()}")
Input parameters
Input parameters needed:
seq: Sequence of the DNA structure (e.g. CGCGAATTCGCG)
seq_comp: Complementary sequence of the given DNA structure (e.g. CGCGAATTCGCG)
traj: Trajectory for a 500ns Drew Dickerson Dodecamer MD simulation (taken from BigNASim)
top: Associated topology for the MD trajectory
# Auxiliary libraries
import os
import shutil
import glob
from pathlib import Path, PurePath
import zipfile
import matplotlib.image as mpimg
import matplotlib.pyplot as plt
import pandas as pd
from IPython.display import Image
import ipywidgets
# Input parameters
seq = "CGCGAATTCGCG"
seq_comp = "CGCGAATTCGCG"
traj = "TRAJ/structure.stripped.nc"
top = "TRAJ/structure.stripped.top"
# Auxiliary lists
grooves = ('majd','majw','mind','minw')
axis_base_pairs = ('inclin','tip','xdisp','ydisp')
base_pair = ('shear','stretch','stagger','buckle','propel','opening')
base_pair_step = ('rise','roll','twist','shift','slide','tilt')
backbone_torsions = ('alphaC', 'alphaW', 'betaC', 'betaW', 'gammaC', 'gammaW', 'deltaC', 'deltaW', \
'epsilC', 'epsilW', 'zetaC', 'zetaW', 'chiC', 'chiW', 'phaseC', 'phaseW')
Running Curves+ and Canal
Curves+ program and its associated Canal tool allow us to extract helical parameters from a DNA MD simulation.
Curves+ is a nucleic acid conformational analysis program which provides both helical and backbone parameters, including a curvilinear axis and parameters relating the position of the bases to this axis. It additionally provides a full analysis of groove widths and depths. Curves+ can also be used to analyse molecular dynamics trajectories. With the help of the accompanying program Canal, it is possible to produce a variety of graphical output including parameter variations along a given structure and time series or histograms of parameter variations during dynamics.
Conformational analysis of nucleic acids revisited: Curves+
R. Lavery, M. Moakher, J. H. Maddocks, D. Petkeviciute, K. Zakrzewska
Nucleic Acids Research, Volume 37, Issue 17, 1 September 2009, Pages 5917–5929
https://doi.org/10.1093/nar/gkp608
CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structure.
C. Blanchet, M. Pasi, K. Zakrzewska, R. Lavery
Nucleic Acids Research, Volume 39, Issue suppl_2, 1 July 2011, Pages W68–W73
https://doi.org/10.1093/nar/gkr316
http://curvesplus.bsc.es
Building Blocks used:
The extraction of helical parameters is then done in two steps:
Step 1: Curves+: Reading input MD trajectory and analysing helical parameters.
Step 2: Canal: Taking Curves+ output and generating time series and/or histograms of parameter variations during dynamics.
Step 1: Curves+
Curves+ program needs a trajectory and its associated topology, and a couple of ranges, informing about the numeration of the two DNA strands: s1range and s2range.
from biobb_dna.curvesplus.biobb_curves import biobb_curves
curves_out_lis = "curves.out.lis"
curves_out_cda = "curves.out.cda"
prop = {
's1range' : '1:12',
's2range' : '24:13',
# uncomment when running in google colab
# 'stdlib_path': '.curvesplus/standard'
}
biobb_curves(
input_struc_path=traj,
input_top_path=top,
output_lis_path=curves_out_lis,
output_cda_path=curves_out_cda,
properties=prop
)
Step 2: Canal
Canal program needs the output of the previous Curves+ execution, and is able to produce time series (series property) and histograms (histo property) for the parameter variations during dynamics.
from biobb_dna.curvesplus.biobb_canal import biobb_canal
canal_out = "canal.out.zip"
prop = {
'series' : True,
'histo' : True
}
biobb_canal(
input_cda_file=curves_out_cda,
input_lis_file=curves_out_lis,
output_zip_path=canal_out,
properties=prop
)
Extracting Canal results in a temporary folder
canal_dir = "canal_out"
if Path(canal_dir).exists(): shutil.rmtree(canal_dir)
os.mkdir(canal_dir)
with zipfile.ZipFile(canal_out, 'r') as zip_ref:
zip_ref.extractall(canal_dir)
Extracting Average Helical Parameters
Average helical parameter values can be computed from the output of Curves+/Canal execution.
The helical parameters can be divided in 5 main blocks:
Helical Base Pair Step (Inter Base Pair) Parameters
Translational (Shift, Slide, Rise) and rotational (Tilt, Roll, Twist) parameters related to a dinucleotide Inter-Base Pair (Base Pair Step).
Shift: Translation around the X-axis.
Slide: Translation around the Y-axis.
Rise: Translation around the Z-axis.
Tilt: Rotation around the X-axis.
Roll: Rotation around the Y-axis.
Twist: Rotation around the Z-axis.

Building Block used:
dna_averages from biobb_dna.dna.dna_averages
Extracting a particular Helical Parameter: Rise
from biobb_dna.dna.dna_averages import dna_averages
helpar = 'rise'
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_averages_csv_path= helpar+'.averages.csv'
output_averages_jpg_path= helpar+'.averages.jpg'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_averages(
input_ser_path=input_file_path,
output_csv_path=output_averages_csv_path,
output_jpg_path=output_averages_jpg_path,
properties=prop)
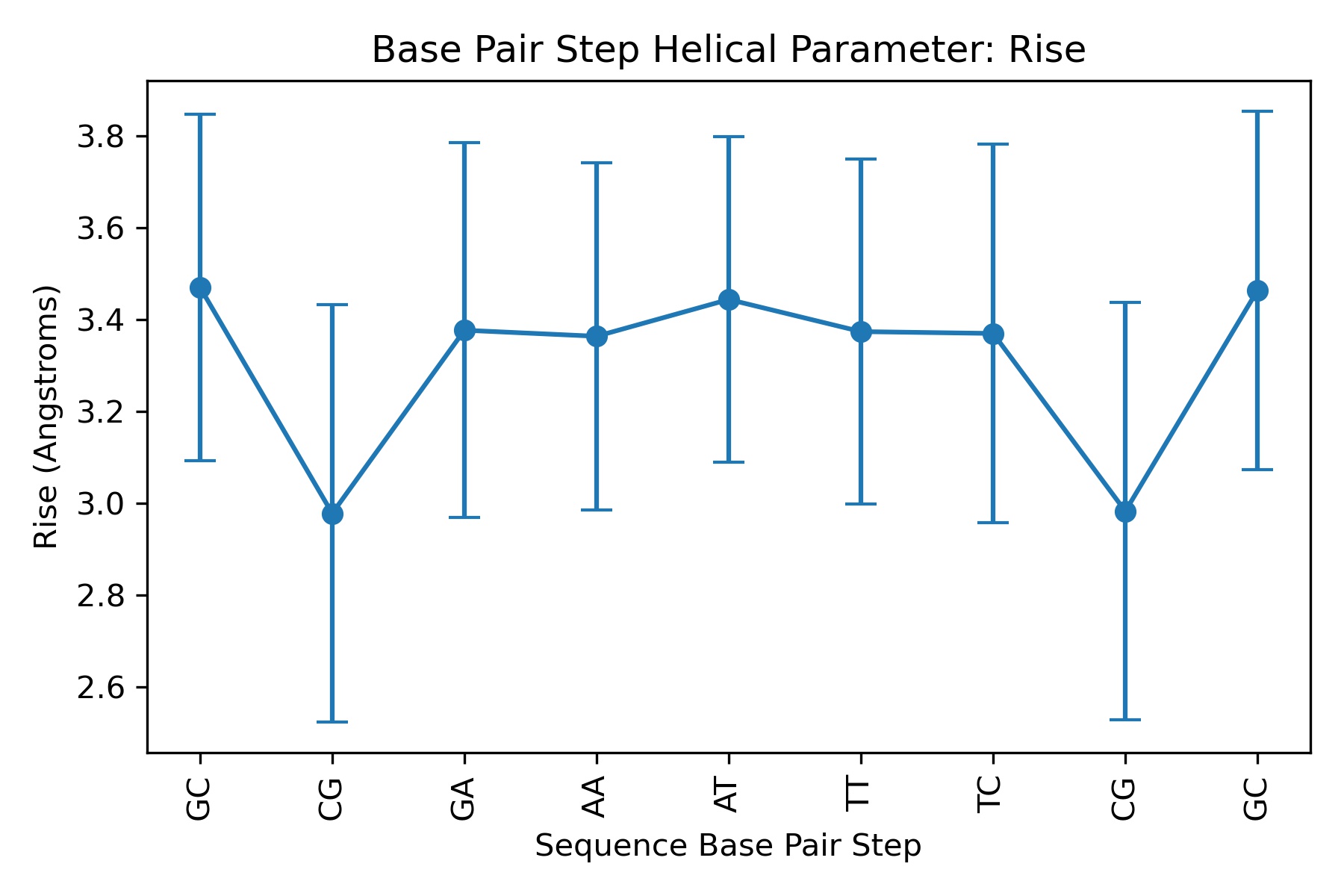
Showing the calculated average values for Rise helical parameter
output_averages_csv_path= helpar+'.averages.csv'
df = pd.read_csv(output_averages_csv_path)
df
| Base Pair Step | mean | std | |
|---|---|---|---|
| 0 | GC | 3.470550 | 0.377972 |
| 1 | CG | 2.978038 | 0.454661 |
| 2 | GA | 3.377096 | 0.408705 |
| 3 | AA | 3.363942 | 0.378155 |
| 4 | AT | 3.444138 | 0.355242 |
| 5 | TT | 3.374110 | 0.376290 |
| 6 | TC | 3.370114 | 0.411934 |
| 7 | CG | 2.982740 | 0.454215 |
| 8 | GC | 3.464096 | 0.390428 |
Plotting the average values for Rise helical parameter
Image(filename=output_averages_jpg_path,width = 600)
Computing average values from all base-pair step parameters
from biobb_dna.dna.dna_averages import dna_averages
for helpar in base_pair_step:
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_averages_csv_path= helpar+'.averages.csv'
output_averages_jpg_path= helpar+'.averages.jpg'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_averages(
input_ser_path=input_file_path,
output_csv_path=output_averages_csv_path,
output_jpg_path=output_averages_jpg_path,
properties=prop)
Showing the calculated average values for all base-pair step helical parameters
for helpar in base_pair_step:
output_averages_csv_path= helpar+'.averages.csv'
df = pd.read_csv(output_averages_csv_path)
print("Helical Parameter: " + helpar)
print(df)
print("---------\n")
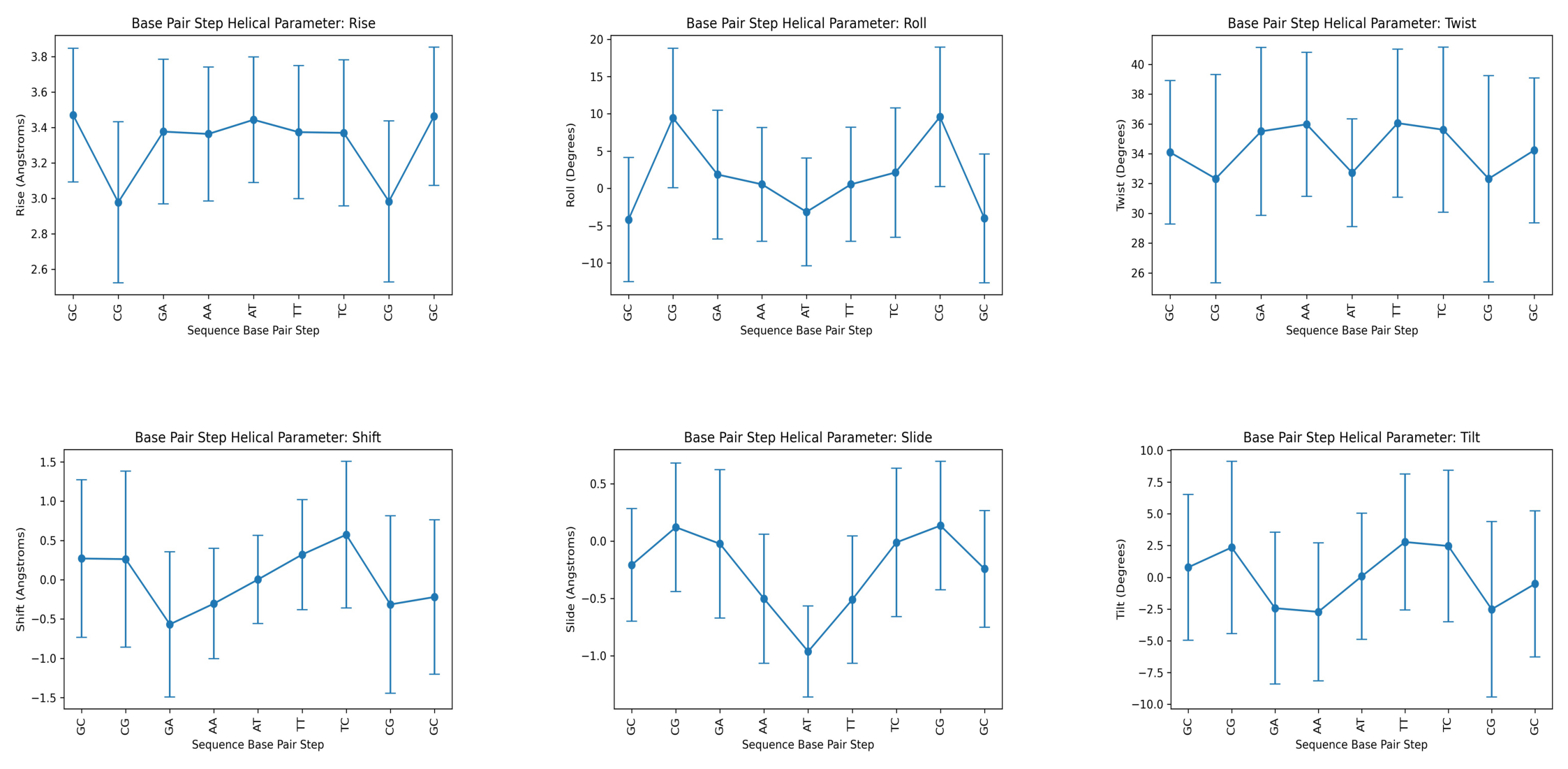
Helical Parameter: rise
Base Pair Step mean std
0 GC 3.470550 0.377972
1 CG 2.978038 0.454661
2 GA 3.377096 0.408705
3 AA 3.363942 0.378155
4 AT 3.444138 0.355242
5 TT 3.374110 0.376290
6 TC 3.370114 0.411934
7 CG 2.982740 0.454215
8 GC 3.464096 0.390428
---------
Helical Parameter: roll
Base Pair Step mean std
0 GC -4.192414 8.331507
1 CG 9.446606 9.363419
2 GA 1.852456 8.644153
3 AA 0.536582 7.624018
4 AT -3.163010 7.237972
5 TT 0.534468 7.656334
6 TC 2.127450 8.682155
7 CG 9.585186 9.365877
8 GC -4.021720 8.647046
---------
Helical Parameter: twist
Base Pair Step mean std
0 GC 34.088546 4.816438
1 CG 32.326028 6.989191
2 GA 35.500510 5.637390
3 AA 35.972860 4.838027
4 AT 32.721506 3.618198
5 TT 36.053014 4.974014
6 TC 35.610722 5.545855
7 CG 32.319386 6.922376
8 GC 34.228190 4.863295
---------
Helical Parameter: shift
Base Pair Step mean std
0 GC 0.269898 1.002226
1 CG 0.261994 1.120109
2 GA -0.568252 0.924852
3 AA -0.303916 0.701930
4 AT 0.003008 0.560834
5 TT 0.320328 0.700562
6 TC 0.573412 0.934916
7 CG -0.315024 1.130856
8 GC -0.220348 0.982379
---------
Helical Parameter: slide
Base Pair Step mean std
0 GC -0.207536 0.492641
1 CG 0.121372 0.560931
2 GA -0.023312 0.648352
3 AA -0.502846 0.563088
4 AT -0.963374 0.396783
5 TT -0.510682 0.556494
6 TC -0.011398 0.648248
7 CG 0.135720 0.561489
8 GC -0.242346 0.509866
---------
Helical Parameter: tilt
Base Pair Step mean std
0 GC 0.788582 5.737113
1 CG 2.352174 6.790473
2 GA -2.433898 5.987960
3 AA -2.723502 5.435644
4 AT 0.072902 4.969859
5 TT 2.780530 5.351470
6 TC 2.466076 5.965965
7 CG -2.520270 6.909690
8 GC -0.519486 5.749269
---------
Plotting the average values for all base-pair step helical parameters
images = []
for helpar in base_pair_step:
images.append(helpar + '.averages.jpg')
f, axarr = plt.subplots(2, 3, figsize=(40, 20))
for i, image in enumerate(images):
y = i%3
x = int(i/3)
img = mpimg.imread(image)
axarr[x,y].imshow(img, aspect='auto')
axarr[x,y].axis('off')
plt.show()
Helical Base Pair (Intra Base Pair) Parameters
Translational (Shear, Stretch, Stagger) and rotational (Buckle, Propeller, Opening) parameters related to a dinucleotide Intra-Base Pair.
Shear: Translation around the X-axis.
Stretch: Translation around the Y-axis.
Stagger: Translation around the Z-axis.
Buckle: Rotation around the X-axis.
Propeller: Rotation around the Y-axis.
Opening: Rotation around the Z-axis.

Building Block used:
dna_averages from biobb_dna.dna.dna_averages
Computing average values from all base-pair parameters
from biobb_dna.dna.dna_averages import dna_averages
for helpar in base_pair:
#input_file_path = canal_out + "_" + helpar + ".ser"
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_averages_csv_path= helpar+'.averages.csv'
output_averages_jpg_path= helpar+'.averages.jpg'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_averages(
input_ser_path=input_file_path,
output_csv_path=output_averages_csv_path,
output_jpg_path=output_averages_jpg_path,
properties=prop)
Showing the calculated average values for all base-pair helical parameters
for helpar in base_pair:
output_averages_csv_path= helpar+'.averages.csv'
df = pd.read_csv(output_averages_csv_path)
print("Helical Parameter: " + helpar)
print(df)
print("---------\n")
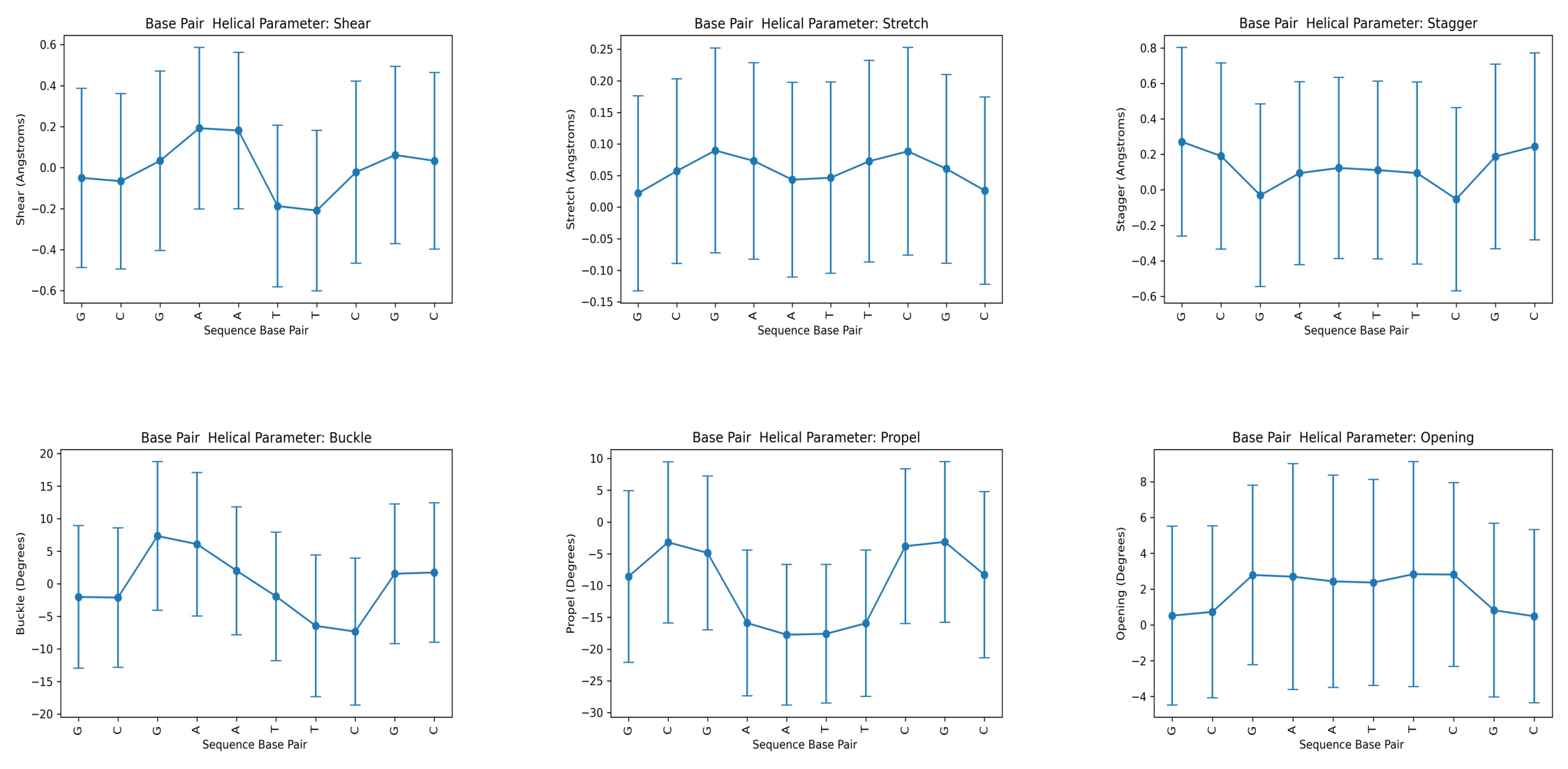
Helical Parameter: shear
Base Pair mean std
0 G -0.049992 0.437419
1 C -0.066120 0.428450
2 G 0.033414 0.438027
3 A 0.192408 0.393915
4 A 0.181268 0.382112
5 T -0.187710 0.394140
6 T -0.209416 0.391508
7 C -0.021976 0.444251
8 G 0.061500 0.432066
9 C 0.033314 0.430674
---------
Helical Parameter: stretch
Base Pair mean std
0 G 0.021910 0.154373
1 C 0.057128 0.146271
2 G 0.089684 0.162121
3 A 0.073182 0.155722
4 A 0.043370 0.154155
5 T 0.046586 0.151565
6 T 0.072776 0.159554
7 C 0.088406 0.164430
8 G 0.060682 0.149506
9 C 0.026160 0.148136
---------
Helical Parameter: stagger
Base Pair mean std
0 G 0.270900 0.531987
1 C 0.190716 0.524902
2 G -0.030574 0.514709
3 A 0.094458 0.515384
4 A 0.123512 0.511518
5 T 0.111454 0.501179
6 T 0.094714 0.513247
7 C -0.052344 0.516656
8 G 0.187962 0.520935
9 C 0.245116 0.527218
---------
Helical Parameter: buckle
Base Pair mean std
0 G -2.011124 10.957743
1 C -2.108248 10.706547
2 G 7.351526 11.411534
3 A 6.094836 11.016860
4 A 2.021262 9.824963
5 T -1.932570 9.869768
6 T -6.454566 10.888373
7 C -7.332888 11.269869
8 G 1.551400 10.722523
9 C 1.740906 10.696502
---------
Helical Parameter: propel
Base Pair mean std
0 G -8.589338 13.487527
1 C -3.200962 12.673929
2 G -4.877606 12.107475
3 A -15.895916 11.463353
4 A -17.740540 11.053182
5 T -17.600944 10.901201
6 T -15.934650 11.522282
7 C -3.809752 12.151864
8 G -3.145238 12.632066
9 C -8.300600 13.082747
---------
Helical Parameter: opening
Base Pair mean std
0 G 0.519980 4.990688
1 C 0.727574 4.801118
2 G 2.788520 5.011118
3 A 2.696966 6.306307
4 A 2.434002 5.929345
5 T 2.367904 5.751322
6 T 2.837478 6.282682
7 C 2.813492 5.139189
8 G 0.822494 4.850359
9 C 0.483850 4.838646
---------
Plotting the average values for all base-pair helical parameters
images = []
for helpar in base_pair:
images.append(helpar + '.averages.jpg')
f, axarr = plt.subplots(2, 3, figsize=(40, 20))
for i, image in enumerate(images):
y = i%3
x = int(i/3)
img = mpimg.imread(image)
axarr[x,y].imshow(img, aspect='auto')
axarr[x,y].axis('off')
plt.show()
Axis Base Pair Parameters
Translational (x/y-displacement) and rotational (inclination, tip) parameters related to a dinucleotide Base Pair.
X-displacement: Translation around the X-axis.
Y-displacement: Translation around the Y-axis.
Inclination: Rotation around the X-axis.
Tip: Rotation around the Y-axis.

Building Block used:
dna_averages from biobb_dna.dna.averages
Computing average values from all Axis base-pair parameters
from biobb_dna.dna.dna_averages import dna_averages
for helpar in axis_base_pairs:
#input_file_path = canal_out + "_" + helpar + ".ser"
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_averages_csv_path= helpar+'.averages.csv'
output_averages_jpg_path= helpar+'.averages.jpg'
prop = {
'helpar_name': helpar,
'sequence': seq,
# 'seqpos': [4,3]
}
dna_averages(
input_ser_path=input_file_path,
output_csv_path=output_averages_csv_path,
output_jpg_path=output_averages_jpg_path,
properties=prop)
Showing the calculated average values for all Axis base-pair helical parameters
for helpar in axis_base_pairs:
output_averages_csv_path= helpar+'.averages.csv'
df = pd.read_csv(output_averages_csv_path)
print("Helical Parameter: " + helpar)
print(df)
print("---------\n")
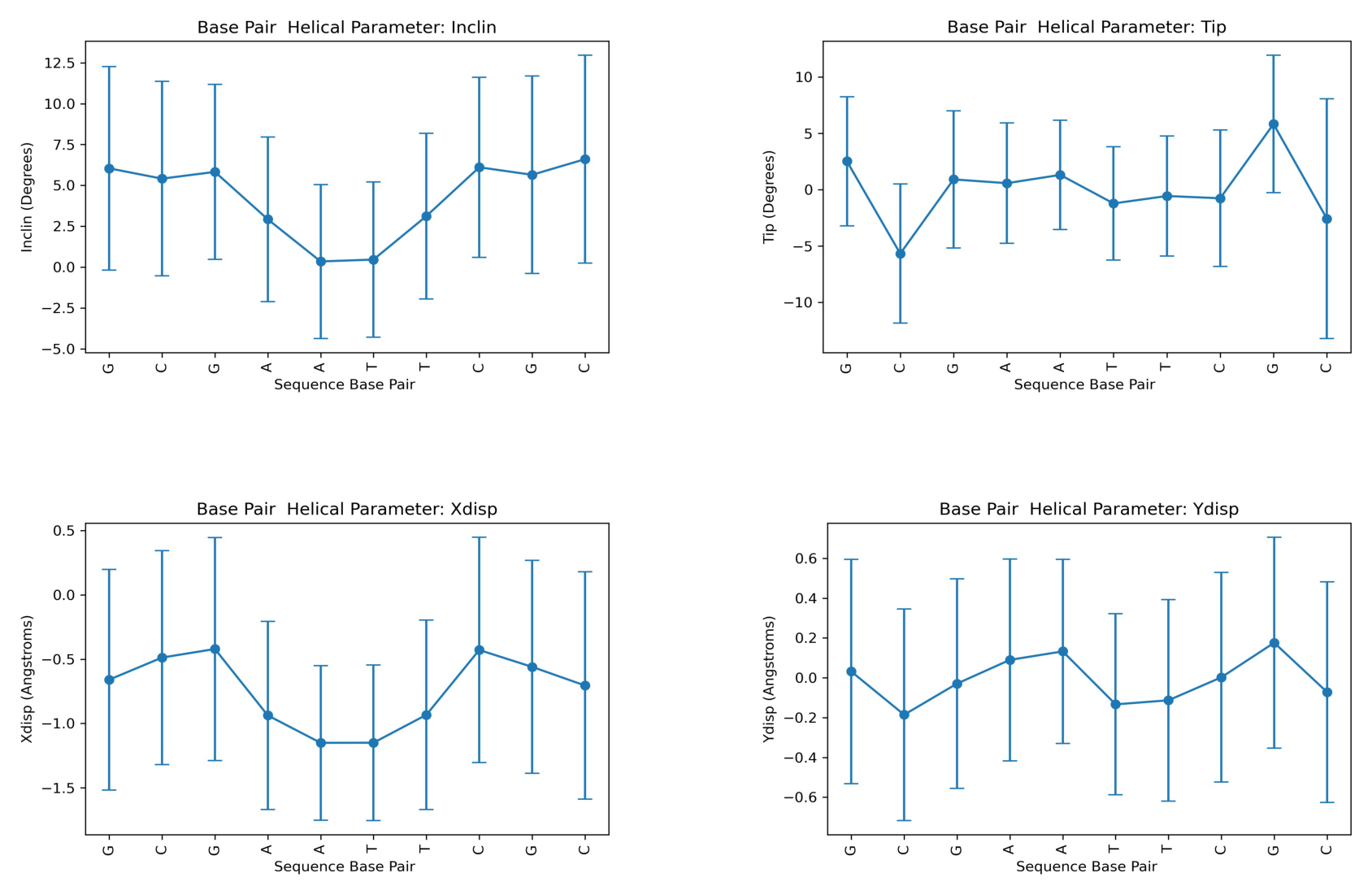
Helical Parameter: inclin
Base Pair mean std
0 G 6.041284 6.230831
1 C 5.410818 5.944490
2 G 5.825528 5.359376
3 A 2.924440 5.031021
4 A 0.346356 4.709278
5 T 0.460490 4.744693
6 T 3.120780 5.069334
7 C 6.106598 5.504673
8 G 5.647446 6.043921
9 C 6.601924 6.358550
---------
Helical Parameter: tip
Base Pair mean std
0 G 2.512608 5.728359
1 C -5.681794 6.177066
2 G 0.907828 6.084601
3 A 0.569286 5.347552
4 A 1.306582 4.845978
5 T -1.227768 5.019692
6 T -0.568284 5.338846
7 C -0.767708 6.048420
8 G 5.821194 6.102313
9 C -2.578986 10.627202
---------
Helical Parameter: xdisp
Base Pair mean std
0 G -0.659664 0.857688
1 C -0.487874 0.832502
2 G -0.421234 0.867164
3 A -0.937412 0.731117
4 A -1.150654 0.600304
5 T -1.149698 0.604352
6 T -0.933022 0.736415
7 C -0.428964 0.875973
8 G -0.560260 0.827803
9 C -0.704972 0.883845
---------
Helical Parameter: ydisp
Base Pair mean std
0 G 0.031522 0.563973
1 C -0.185588 0.531173
2 G -0.029302 0.526326
3 A 0.089948 0.507185
4 A 0.132712 0.462237
5 T -0.133432 0.454954
6 T -0.113044 0.506105
7 C 0.002278 0.526475
8 G 0.176116 0.529698
9 C -0.071754 0.554503
---------
Plotting the average values for all Axis base-pair helical parameters
images = []
for helpar in axis_base_pairs:
images.append(helpar + '.averages.jpg')
f, axarr = plt.subplots(2, 2, figsize=(30, 20))
for i, image in enumerate(images):
y = i%2
x = int(i/2)
img = mpimg.imread(image)
axarr[x,y].imshow(img, aspect='auto')
axarr[x,y].axis('off')
plt.show()
Grooves
Nucleic Acid Structure’s strand backbones appear closer together on one side of the helix than on the other. This creates a Major groove (where backbones are far apart) and a Minor groove (where backbones are close together). Depth and width of these grooves can be mesured giving information about the different conformations that the nucleic acid structure can achieve.
Major Groove Width.
Major Groove Depth.
Minor Groove Width.
Minor Groove Depth.

Building Block used:
dna_averages from biobb_dna.dna.dna_averages
Computing average values from all Grooves parameters
from biobb_dna.dna.dna_averages import dna_averages
for helpar in grooves:
#input_file_path = canal_out + "_" + helpar + ".ser"
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_averages_csv_path= helpar+'.averages.csv'
output_averages_jpg_path= helpar+'.averages.jpg'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_averages(
input_ser_path=input_file_path,
output_csv_path=output_averages_csv_path,
output_jpg_path=output_averages_jpg_path,
properties=prop)
Showing the calculated average values for all Grooves parameters
for helpar in grooves:
output_averages_csv_path= helpar+'.averages.csv'
df = pd.read_csv(output_averages_csv_path)
print("Helical Parameter: " + helpar)
print(df)
print("---------\n")
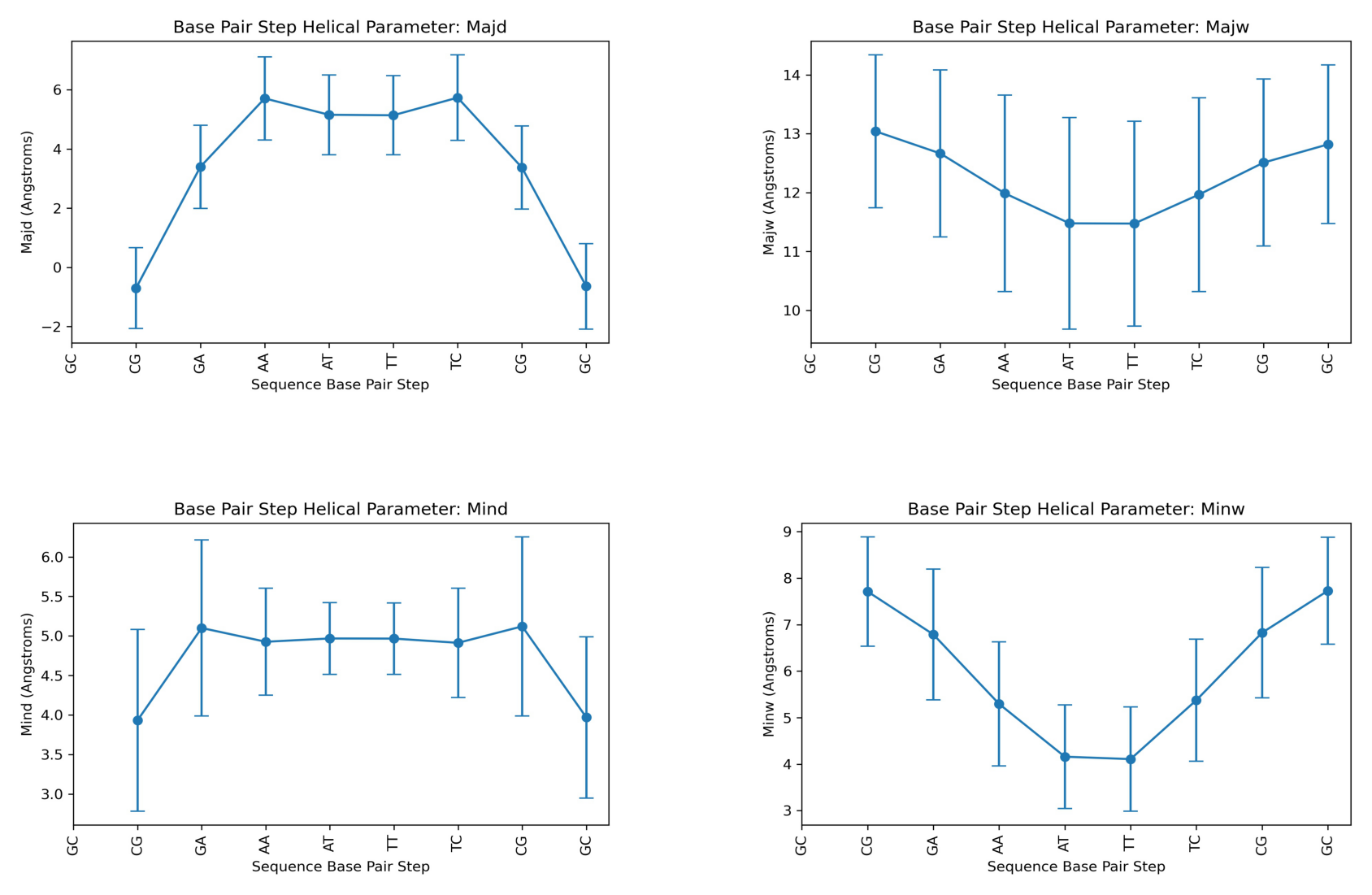
Helical Parameter: majd
Base Pair Step mean std
0 GC NaN NaN
1 CG -0.703077 1.362241
2 GA 3.396398 1.402178
3 AA 5.710123 1.406443
4 AT 5.156143 1.346752
5 TT 5.139702 1.337523
6 TC 5.735556 1.447770
7 CG 3.371511 1.402279
8 GC -0.644086 1.444480
---------
Helical Parameter: majw
Base Pair Step mean std
0 GC NaN NaN
1 CG 13.040615 1.300370
2 GA 12.664800 1.419105
3 AA 11.986143 1.670021
4 AT 11.476450 1.797558
5 TT 11.472435 1.740485
6 TC 11.964836 1.648435
7 CG 12.508447 1.419017
8 GC 12.819785 1.347786
---------
Helical Parameter: mind
Base Pair Step mean std
0 GC NaN NaN
1 CG 3.932224 1.148757
2 GA 5.100660 1.114656
3 AA 4.926210 0.677415
4 AT 4.968002 0.453657
5 TT 4.966188 0.452291
6 TC 4.912856 0.690885
7 CG 5.121034 1.132328
8 GC 3.969312 1.021299
---------
Helical Parameter: minw
Base Pair Step mean std
0 GC NaN NaN
1 CG 7.713347 1.175525
2 GA 6.789962 1.405118
3 AA 5.296306 1.332686
4 AT 4.160776 1.113765
5 TT 4.109198 1.119448
6 TC 5.376128 1.314328
7 CG 6.827374 1.402630
8 GC 7.729192 1.148585
---------
Plotting the average values for all Grooves helical parameters
images = []
for helpar in grooves:
images.append(helpar + '.averages.jpg')
f, axarr = plt.subplots(2, 2, figsize=(30, 20))
for i, image in enumerate(images):
y = i%2
x = int(i/2)
img = mpimg.imread(image)
axarr[x,y].imshow(img, aspect='auto')
axarr[x,y].axis('off')
plt.show()
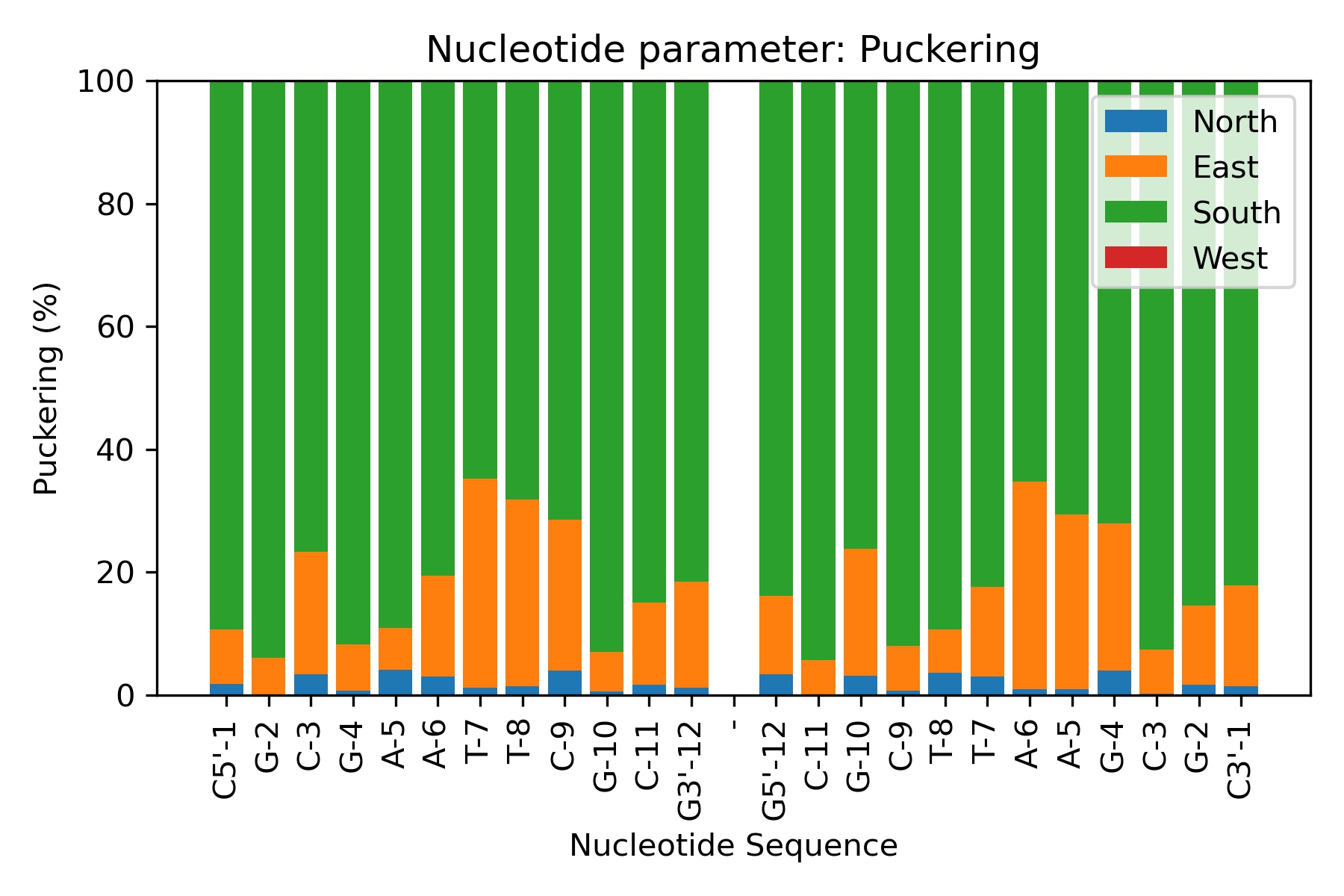
Backbone Torsions
The three major elements of flexibility in the backbone are:
-
Sugar Puckering annotation is done by dividing the pseudo-rotational circle in four equivalent sections:
North: 315:45º
East: 45:135º
South: 135:225º
West: 225:315º
These four conformations are those dominating sugar conformational space, in agreement with all available experimental data.
-
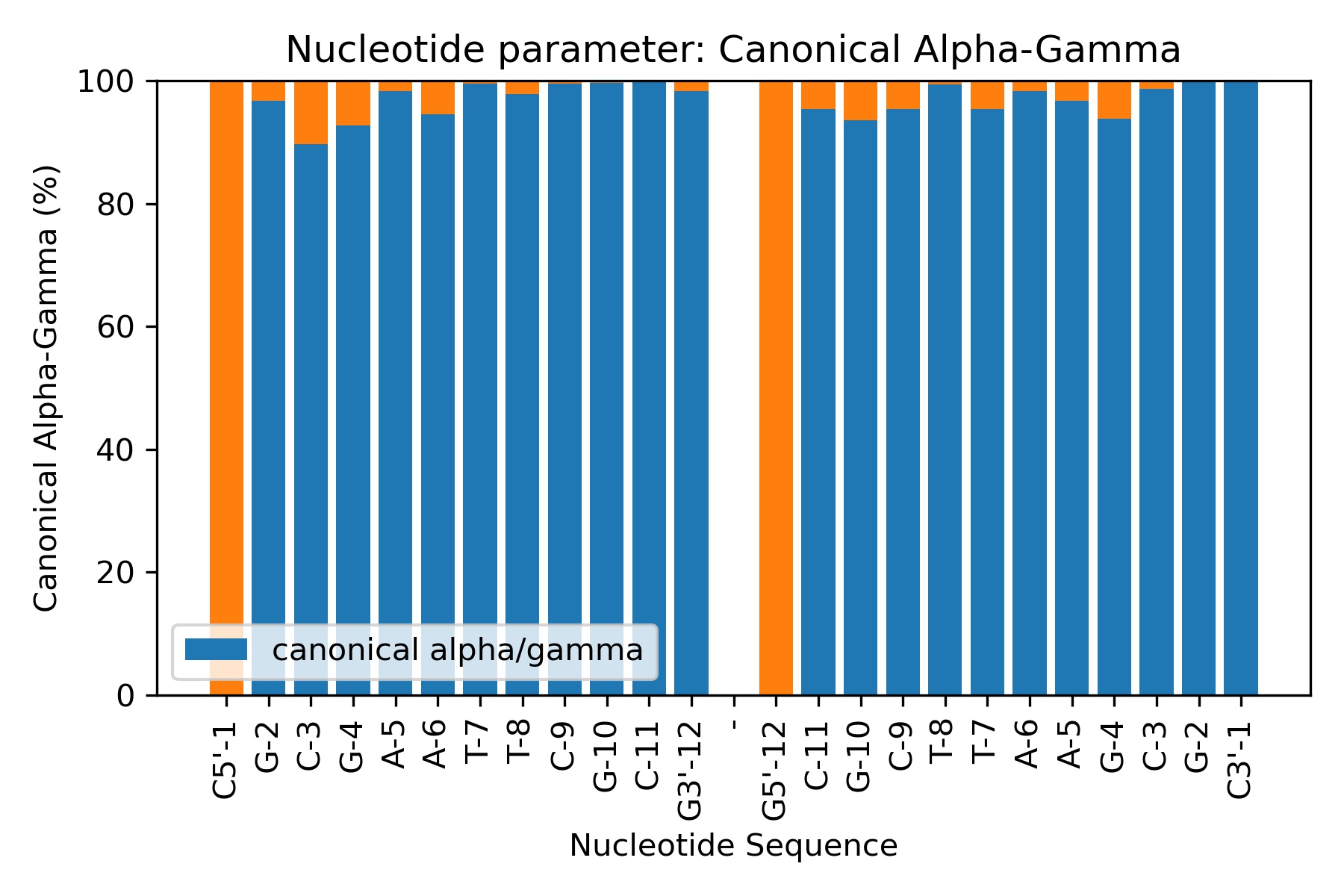
Rotations around α/γ torsions generate non-canonical local conformations leading to a reduced twist and they have been reported as being important in the formation of several protein-DNA complexes.
-
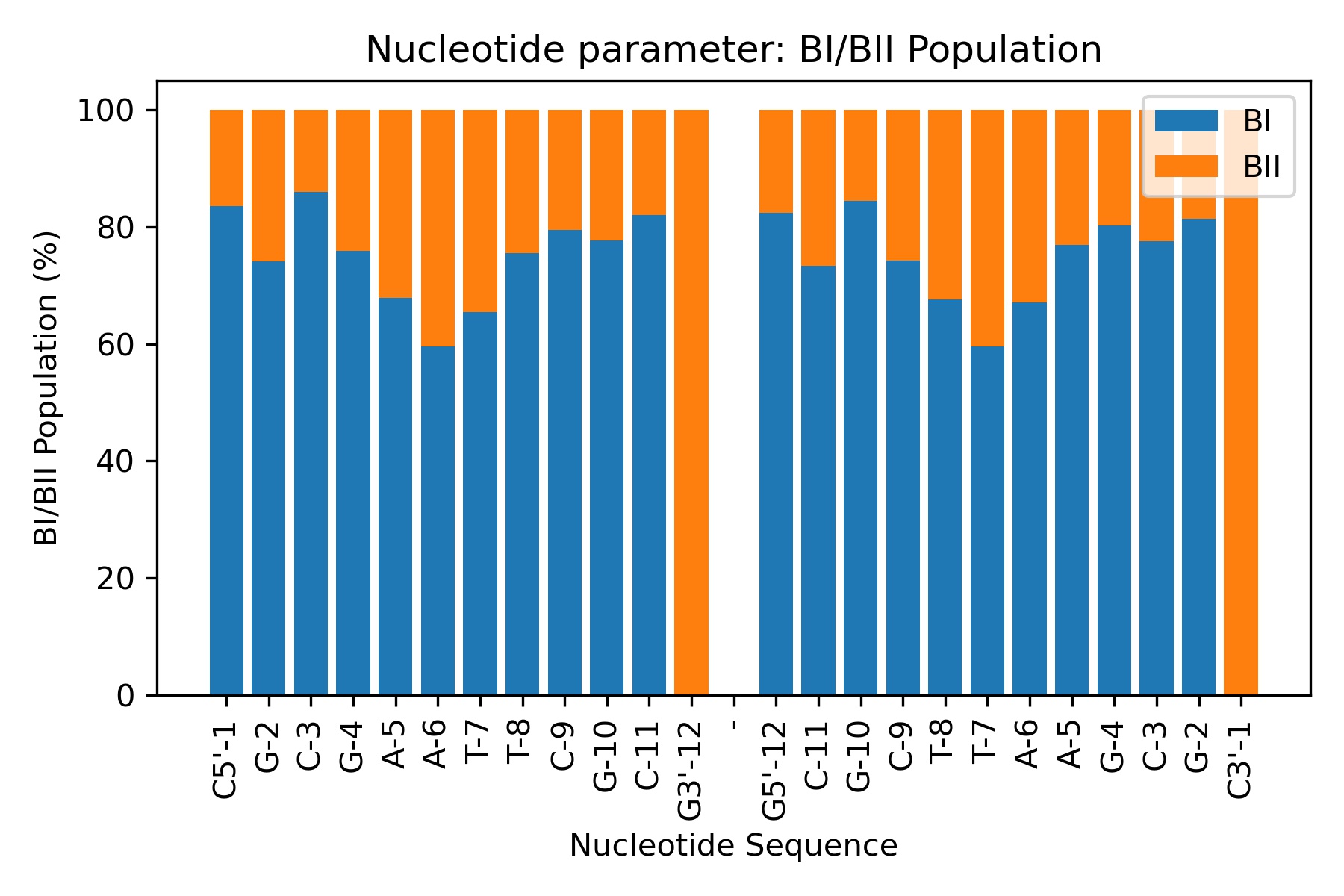
The concerted rotation around ζ/ε torsions generates two major conformers: BI and BII, which are experimentally known to co-exist in a ratio around 80%:20% (BI:BII) in B-DNA.

|

|

|
|
Sugar Puckering |
Canonical Alpha/Gamma |
BI/BII population |
Building Blocks used:
puckering from biobb_dna.backbone.puckering
canonicalag from biobb_dna.backbone.canonicalag
bipopulations from biobb_dna.backbone.bipopulations
Sugar Puckering
Computing average values
from biobb_dna.backbone.puckering import puckering
canal_phaseC = "canal_out/canal_output_phaseC.ser"
canal_phaseW = "canal_out/canal_output_phaseW.ser"
output_puckering_csv_path = 'puckering.averages.csv'
output_puckering_jpg_path = 'puckering.averages.jpg'
prop = {
'sequence': seq
}
puckering(
input_phaseC_path=canal_phaseC,
input_phaseW_path=canal_phaseW,
output_csv_path=output_puckering_csv_path,
output_jpg_path=output_puckering_jpg_path,
properties=prop)
Showing the calculated average values
df = pd.read_csv(output_puckering_csv_path)
df
| Nucleotide | North | East | West | South | |
|---|---|---|---|---|---|
| 0 | C5'-1 | 1.82 | 8.84 | 0.02 | 89.32 |
| 1 | G-2 | 0.10 | 5.92 | 0.02 | 93.96 |
| 2 | C-3 | 3.36 | 20.02 | 0.00 | 76.60 |
| 3 | G-4 | 0.76 | 7.52 | 0.00 | 91.72 |
| 4 | A-5 | 4.16 | 6.72 | 0.00 | 89.10 |
| 5 | A-6 | 3.04 | 16.44 | 0.00 | 80.50 |
| 6 | T-7 | 1.16 | 34.06 | 0.00 | 64.76 |
| 7 | T-8 | 1.50 | 30.36 | 0.00 | 68.10 |
| 8 | C-9 | 4.00 | 24.56 | 0.00 | 71.44 |
| 9 | G-10 | 0.60 | 6.40 | 0.02 | 92.98 |
| 10 | C-11 | 1.72 | 13.38 | 0.00 | 84.90 |
| 11 | G3'-12 | 1.18 | 17.28 | 0.04 | 81.50 |
| 12 | - | 0.00 | 0.00 | 0.00 | 0.00 |
| 13 | G5'-12 | 3.44 | 12.72 | 0.02 | 83.80 |
| 14 | C-11 | 0.16 | 5.56 | 0.00 | 94.24 |
| 15 | G-10 | 3.14 | 20.70 | 0.00 | 76.14 |
| 16 | C-9 | 0.70 | 7.32 | 0.00 | 91.98 |
| 17 | T-8 | 3.64 | 7.04 | 0.00 | 89.32 |
| 18 | T-7 | 3.02 | 14.58 | 0.02 | 82.38 |
| 19 | A-6 | 0.96 | 33.82 | 0.00 | 65.22 |
| 20 | A-5 | 1.00 | 28.42 | 0.00 | 70.58 |
| 21 | G-4 | 4.00 | 23.96 | 0.02 | 71.98 |
| 22 | C-3 | 0.24 | 7.14 | 0.02 | 92.60 |
| 23 | G-2 | 1.72 | 12.86 | 0.00 | 85.40 |
| 24 | C3'-1 | 1.40 | 16.46 | 0.00 | 82.12 |
Plotting the average values
Image(filename=output_puckering_jpg_path,width = 600)
Canonical Alpha/Gamma
Computing average values
from biobb_dna.backbone.canonicalag import canonicalag
canal_alphaC = "canal_out/canal_output_alphaC.ser"
canal_alphaW = "canal_out/canal_output_alphaW.ser"
canal_gammaC = "canal_out/canal_output_gammaC.ser"
canal_gammaW = "canal_out/canal_output_gammaW.ser"
output_alphagamma_csv_path = 'alphagamma.averages.csv'
output_alphagamma_jpg_path = 'alphagamma.averages.jpg'
prop = {
'sequence': seq
}
canonicalag(
input_alphaC_path=canal_alphaC,
input_alphaW_path=canal_alphaW,
input_gammaC_path=canal_gammaC,
input_gammaW_path=canal_gammaW,
output_csv_path=output_alphagamma_csv_path,
output_jpg_path=output_alphagamma_jpg_path,
properties=prop)
Showing the calculated average values
df = pd.read_csv(output_alphagamma_csv_path)
df
| Nucleotide | Canonical alpha/gamma | |
|---|---|---|
| 0 | C5'-1 | 0.00 |
| 1 | G-2 | 96.76 |
| 2 | C-3 | 89.66 |
| 3 | G-4 | 92.70 |
| 4 | A-5 | 98.30 |
| 5 | A-6 | 94.52 |
| 6 | T-7 | 99.56 |
| 7 | T-8 | 97.86 |
| 8 | C-9 | 99.54 |
| 9 | G-10 | 99.64 |
| 10 | C-11 | 99.72 |
| 11 | G3'-12 | 98.24 |
| 12 | - | 0.00 |
| 13 | G5'-12 | 0.00 |
| 14 | C-11 | 95.42 |
| 15 | G-10 | 93.56 |
| 16 | C-9 | 95.40 |
| 17 | T-8 | 99.38 |
| 18 | T-7 | 95.36 |
| 19 | A-6 | 98.28 |
| 20 | A-5 | 96.70 |
| 21 | G-4 | 93.86 |
| 22 | C-3 | 98.70 |
| 23 | G-2 | 100.00 |
| 24 | C3'-1 | 99.78 |
Plotting the average values
Image(filename=output_alphagamma_jpg_path,width = 600)
BI/BII Population
Computing average values
from biobb_dna.backbone.bipopulations import bipopulations
canal_epsilC = "canal_out/canal_output_epsilC.ser"
canal_epsilW = "canal_out/canal_output_epsilW.ser"
canal_zetaC = "canal_out/canal_output_zetaC.ser"
canal_zetaW = "canal_out/canal_output_zetaW.ser"
output_bIbII_csv_path = 'bIbII.averages.csv'
output_bIbII_jpg_path = 'bIbII.averages.jpg'
prop = {
'sequence': seq
}
bipopulations(
input_epsilC_path=canal_epsilC,
input_epsilW_path=canal_epsilW,
input_zetaC_path=canal_zetaC,
input_zetaW_path=canal_zetaW,
output_csv_path=output_bIbII_csv_path,
output_jpg_path=output_bIbII_jpg_path,
properties=prop)
Showing the calculated average values
df = pd.read_csv(output_bIbII_csv_path)
df
| Nucleotide | BI population | BII population | |
|---|---|---|---|
| 0 | C5'-1 | 83.523295 | 16.476705 |
| 1 | G-2 | 74.085183 | 25.914817 |
| 2 | C-3 | 85.982803 | 14.017197 |
| 3 | G-4 | 75.924815 | 24.075185 |
| 4 | A-5 | 67.846431 | 32.153569 |
| 5 | A-6 | 59.588082 | 40.411918 |
| 6 | T-7 | 65.406919 | 34.593081 |
| 7 | T-8 | 75.524895 | 24.475105 |
| 8 | C-9 | 79.504099 | 20.495901 |
| 9 | G-10 | 77.644471 | 22.355529 |
| 10 | C-11 | 82.003599 | 17.996401 |
| 11 | G3'-12 | 0.000000 | 100.000000 |
| 12 | - | 0.000000 | 100.000000 |
| 13 | G5'-12 | 82.443511 | 17.556489 |
| 14 | C-11 | 73.305339 | 26.694661 |
| 15 | G-10 | 84.483103 | 15.516897 |
| 16 | C-9 | 74.305139 | 25.694861 |
| 17 | T-8 | 67.606479 | 32.393521 |
| 18 | T-7 | 59.568086 | 40.431914 |
| 19 | A-6 | 67.126575 | 32.873425 |
| 20 | A-5 | 76.984603 | 23.015397 |
| 21 | G-4 | 80.243951 | 19.756049 |
| 22 | C-3 | 77.604479 | 22.395521 |
| 23 | G-2 | 81.443711 | 18.556289 |
| 24 | C3'-1 | 0.000000 | 100.000000 |
Plotting the average values
Image(filename=output_bIbII_jpg_path,width = 600)
Extracting Time series Helical Parameters
Time series values for the set of helical parameters can be also extracted from the output of Curves+/Canal execution on Molecular Dynamics Trajectories. The helical parameters can be divided in the same 5 main blocks previously introduced:
Helical Base Pair Step (Inter-Base Pair) Helical Parameters
Helical Base Pair (Intra-Base Pair) Helical Parameters
Axis Base Pair
Grooves
Backbone Torsions
Building Block used:
dna_timeseries from biobb_dna.dna.dna_timeseries
Extracting a particular Helical Parameter
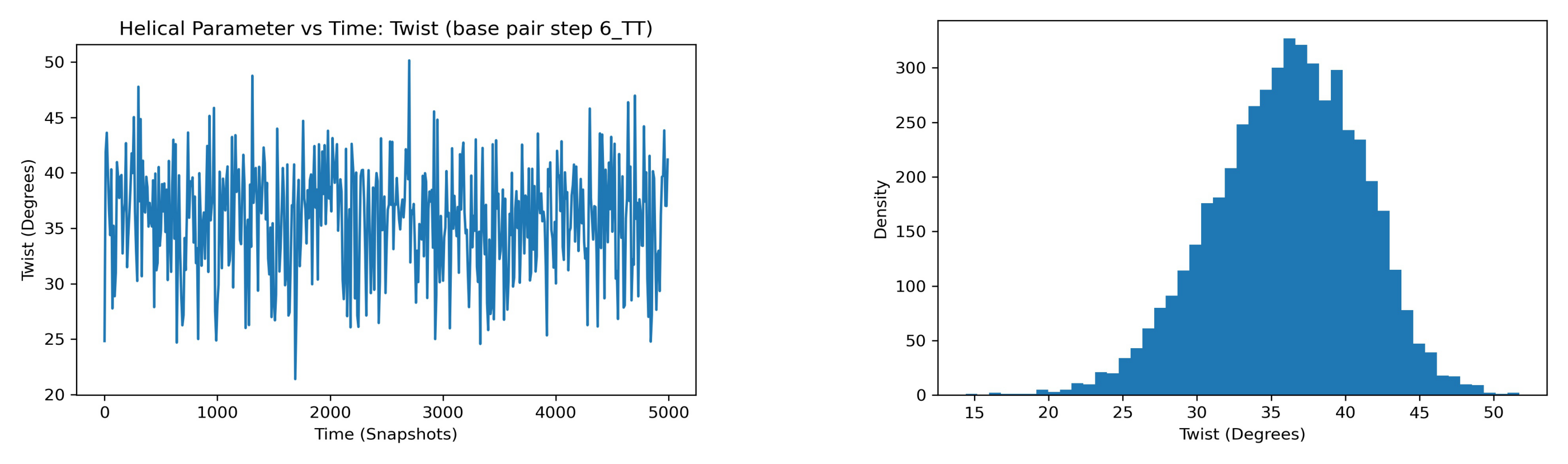
Time series values can be extracted from any of the helical parameters previously introduced. To illustrate the steps needed, the base-pair step helical parameter Twist has been selected. Please note that computing the time series values for a different helical parameter just requires modifying the helpar variable from the next cell.
from biobb_dna.dna.dna_timeseries import dna_timeseries
# Modify the next variable to extract time series values for a different helical parameter
# Possible values are:
# Base Pair Step (Inter Base Pair) Helical Parameters: shift, slide, rise, tilt, roll, twist
# Base Pair (Intra Base Pair) Helical Parameters: shear, stretch, stagger, buckle, propeller, opening
# Axis Parameters: inclin, tip, xdisp, ydisp
# Backbone Torsions Parameters: alphaC, alphaW, betaC, betaW, gammaC, gammaW, deltaC, deltaW,
# epsilC, epsilW, zetaC, zetaW, chiC, chiW, phaseC, phaseW
# Grooves: mind, minw, majd, majw
helpar = "twist" # Modify this variable to extract time series values for a different helical parameter
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_timeseries_file_path = helpar + '.timeseries.zip'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_timeseries(
input_ser_path=input_file_path,
output_zip_path=output_timeseries_file_path,
properties=prop)
Extracting time series results for the selected helical parameter in a temporary folder
timeseries_dir = "timeseries"
if Path(timeseries_dir).exists(): shutil.rmtree(timeseries_dir)
os.mkdir(timeseries_dir)
with zipfile.ZipFile(output_timeseries_file_path, 'r') as zip_ref:
zip_ref.extractall(timeseries_dir)
Finding out all the possible nucleotide / base / base-pair / base-pair steps
Discover all the possible nucleotide / base / base-pair / base-pair steps from the sequence. The unit will depend on the helical parameter being studied.
Select one of them to study the time series values of the helical parameter along the simulation.
helpartimesfiles = glob.glob(timeseries_dir + "/*series*.csv")
helpartimes = []
for file in helpartimesfiles:
new_string = file.replace(timeseries_dir + "/series_" + helpar + "_", "")
new_string = new_string.replace(".csv" , "")
helpartimes.append(new_string)
timesel = ipywidgets.Dropdown(
options=helpartimes,
description='Sel. BPS:',
disabled=False,
)
display(timesel)
Showing the time series values for the selected unit
file_ser = timeseries_dir + "/series_" + helpar + "_" + timesel.value + ".csv"
df = pd.read_csv(file_ser)
df
| Unnamed: 0 | 6_TT | |
|---|---|---|
| 0 | 0 | 24.83 |
| 1 | 1 | 36.78 |
| 2 | 2 | 41.00 |
| 3 | 3 | 33.50 |
| 4 | 4 | 29.68 |
| ... | ... | ... |
| 4995 | 4995 | 42.40 |
| 4996 | 4996 | 44.61 |
| 4997 | 4997 | 39.61 |
| 4998 | 4998 | 36.93 |
| 4999 | 4999 | 36.63 |
5000 rows × 2 columns
file_hist = timeseries_dir + "/hist_" + helpar + "_" + timesel.value + ".csv"
df = pd.read_csv(file_hist)
df
| twist | density | |
|---|---|---|
| 0 | 14.410000 | 1.0 |
| 1 | 15.203617 | 0.0 |
| 2 | 15.997234 | 2.0 |
| 3 | 16.790851 | 1.0 |
| 4 | 17.584468 | 1.0 |
| 5 | 18.378085 | 1.0 |
| 6 | 19.171702 | 5.0 |
| 7 | 19.965319 | 3.0 |
| 8 | 20.758936 | 5.0 |
| 9 | 21.552553 | 11.0 |
| 10 | 22.346170 | 10.0 |
| 11 | 23.139787 | 21.0 |
| 12 | 23.933404 | 20.0 |
| 13 | 24.727021 | 34.0 |
| 14 | 25.520638 | 43.0 |
| 15 | 26.314255 | 61.0 |
| 16 | 27.107872 | 80.0 |
| 17 | 27.901489 | 91.0 |
| 18 | 28.695106 | 114.0 |
| 19 | 29.488723 | 138.0 |
| 20 | 30.282340 | 176.0 |
| 21 | 31.075957 | 181.0 |
| 22 | 31.869574 | 208.0 |
| 23 | 32.663191 | 248.0 |
| 24 | 33.456809 | 265.0 |
| 25 | 34.250426 | 280.0 |
| 26 | 35.044043 | 300.0 |
| 27 | 35.837660 | 327.0 |
| 28 | 36.631277 | 321.0 |
| 29 | 37.424894 | 304.0 |
| 30 | 38.218511 | 270.0 |
| 31 | 39.012128 | 298.0 |
| 32 | 39.805745 | 243.0 |
| 33 | 40.599362 | 234.0 |
| 34 | 41.392979 | 196.0 |
| 35 | 42.186596 | 169.0 |
| 36 | 42.980213 | 115.0 |
| 37 | 43.773830 | 78.0 |
| 38 | 44.567447 | 47.0 |
| 39 | 45.361064 | 39.0 |
| 40 | 46.154681 | 18.0 |
| 41 | 46.948298 | 17.0 |
| 42 | 47.741915 | 10.0 |
| 43 | 48.535532 | 9.0 |
| 44 | 49.329149 | 2.0 |
| 45 | 50.122766 | 1.0 |
| 46 | 50.916383 | 2.0 |
Plotting the time series values for the selected base-pair step
file_ser = timeseries_dir + "/series_" + helpar + "_" + timesel.value + ".jpg"
file_hist = timeseries_dir + "/hist_" + helpar + "_" + timesel.value + ".jpg"
images = []
images.append(file_ser)
images.append(file_hist)
f, axarr = plt.subplots(1, 2, figsize=(50, 15))
for i, image in enumerate(images):
img = mpimg.imread(image)
axarr[i].imshow(img, aspect='auto')
axarr[i].axis('off')
plt.show()
Computing timeseries for all base-pair step parameters
from biobb_dna.dna.dna_timeseries import dna_timeseries
output_timeseries_bps_file_paths = {}
for helpar in base_pair_step:
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_timeseries_bps_file_paths[helpar] = helpar + '.timeseries.zip'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_timeseries(
input_ser_path=input_file_path,
output_zip_path=output_timeseries_bps_file_paths[helpar],
properties=prop)
#if Path(timeseries_dir).exists(): shutil.rmtree(timeseries_dir)
#os.mkdir(timeseries_dir)
for timeseries_zipfile in output_timeseries_bps_file_paths.values():
with zipfile.ZipFile(timeseries_zipfile, 'r') as zip_ref:
zip_ref.extractall(timeseries_dir)
Computing timeseries for all base-pair parameters
from biobb_dna.dna.dna_timeseries import dna_timeseries
output_timeseries_bp_file_paths = {}
for helpar in base_pair:
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_timeseries_bp_file_paths[helpar] = helpar + '.timeseries.zip'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_timeseries(
input_ser_path=input_file_path,
output_zip_path=output_timeseries_bp_file_paths[helpar],
properties=prop)
#if Path(timeseries_dir).exists(): shutil.rmtree(timeseries_dir)
#os.mkdir(timeseries_dir)
for timeseries_zipfile in output_timeseries_bp_file_paths.values():
with zipfile.ZipFile(timeseries_zipfile, 'r') as zip_ref:
zip_ref.extractall(timeseries_dir)
Computing timeseries for all axis parameters
from biobb_dna.dna.dna_timeseries import dna_timeseries
output_timeseries_bp_file_paths = {}
for helpar in axis_base_pairs:
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_timeseries_bp_file_paths[helpar] = helpar + '.timeseries.zip'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_timeseries(
input_ser_path=input_file_path,
output_zip_path=output_timeseries_bp_file_paths[helpar],
properties=prop)
for timeseries_zipfile in output_timeseries_bp_file_paths.values():
with zipfile.ZipFile(timeseries_zipfile, 'r') as zip_ref:
zip_ref.extractall(timeseries_dir)
Computing timeseries for all grooves parameters
from biobb_dna.dna.dna_timeseries import dna_timeseries
output_timeseries_bp_file_paths = {}
for helpar in grooves:
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_timeseries_bp_file_paths[helpar] = helpar + '.timeseries.zip'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_timeseries(
input_ser_path=input_file_path,
output_zip_path=output_timeseries_bp_file_paths[helpar],
properties=prop)
for timeseries_zipfile in output_timeseries_bp_file_paths.values():
with zipfile.ZipFile(timeseries_zipfile, 'r') as zip_ref:
zip_ref.extractall(timeseries_dir)
Computing timeseries for all backbone torsions parameters
from biobb_dna.dna.dna_timeseries import dna_timeseries
output_timeseries_bp_file_paths = {}
for helpar in backbone_torsions:
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_timeseries_bp_file_paths[helpar] = helpar + '.timeseries.zip'
prop = {
'helpar_name': helpar,
'sequence': seq
}
dna_timeseries(
input_ser_path=input_file_path,
output_zip_path=output_timeseries_bp_file_paths[helpar],
properties=prop)
for timeseries_zipfile in output_timeseries_bp_file_paths.values():
with zipfile.ZipFile(timeseries_zipfile, 'r') as zip_ref:
zip_ref.extractall(timeseries_dir)
Stiffness
Molecular stiffness is an elastic force constant associated with helical deformation at the base pair step level and is determined by inversion of the covariance matrix in helical space, which yields stiffness matrices whose diagonal elements provide the stiffness constants associated with pure rotational (twist, roll and tilt) and translational (rise, shift and slide) deformations within the given step.

Building Blocks used:
average_stiffness from biobb_dna.stiffness.average_stiffness
basepair_stiffness from biobb_dna.stiffness.basepair_stiffness
from biobb_dna.stiffness.average_stiffness import average_stiffness
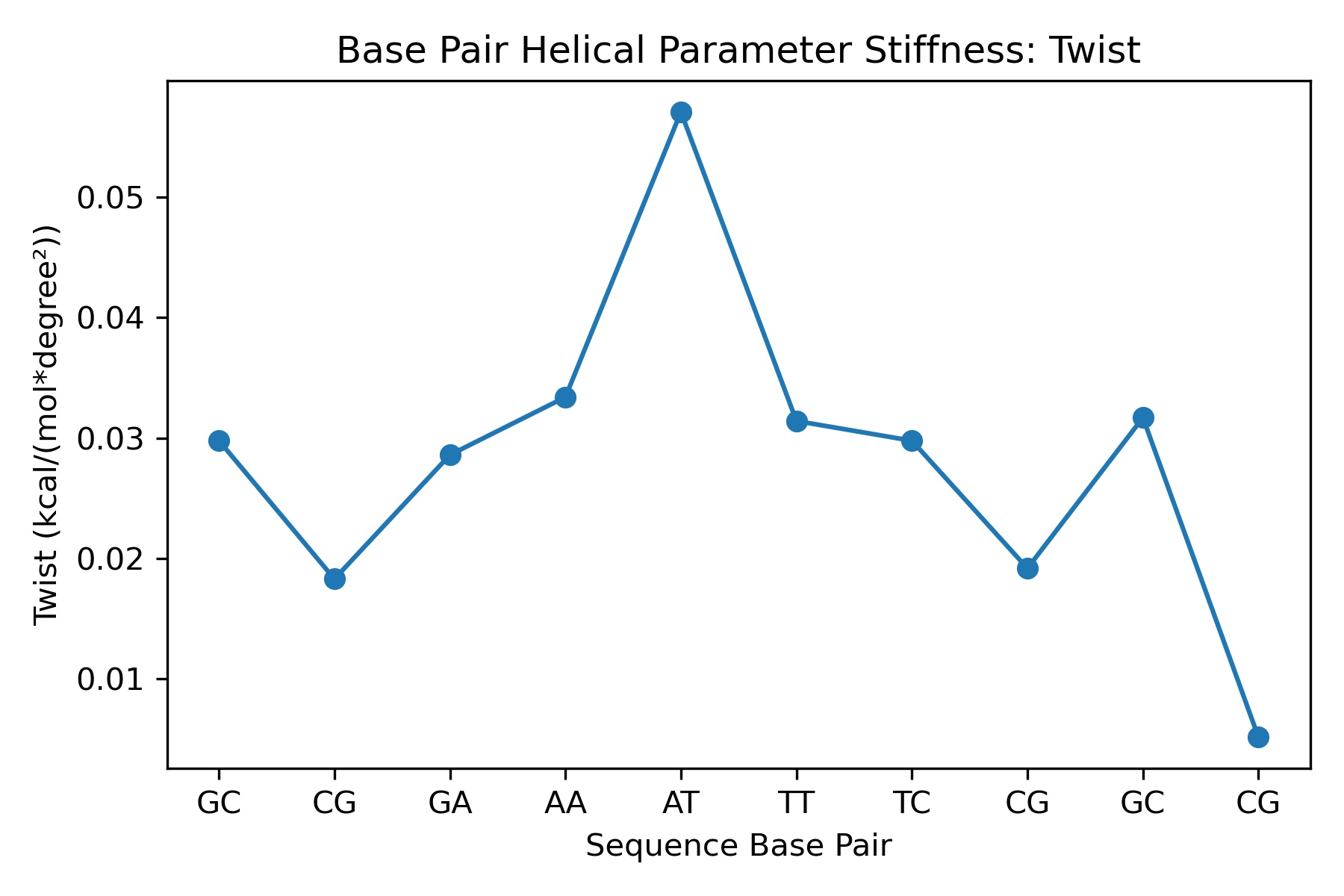
helpar = "twist" # Modify this variable to extract time series values for a different helical parameter
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_stiffness_csv_path = helpar + '.stiffness.csv'
output_stiffness_jpg_path = helpar + '.stiffness.jpg'
prop = {
'sequence' : seq
}
average_stiffness(
input_ser_path=input_file_path,
output_csv_path=output_stiffness_csv_path,
output_jpg_path=output_stiffness_jpg_path,
properties=prop)
df = pd.read_csv(output_stiffness_csv_path)
df
| Unnamed: 0 | twist_stiffness | |
|---|---|---|
| 0 | GC | 0.029784 |
| 1 | CG | 0.018300 |
| 2 | GA | 0.028595 |
| 3 | AA | 0.033391 |
| 4 | AT | 0.057062 |
| 5 | TT | 0.031410 |
| 6 | TC | 0.029772 |
| 7 | CG | 0.019181 |
| 8 | GC | 0.031688 |
| 9 | CG | 0.005167 |
Image(filename=output_stiffness_jpg_path,width = 600)
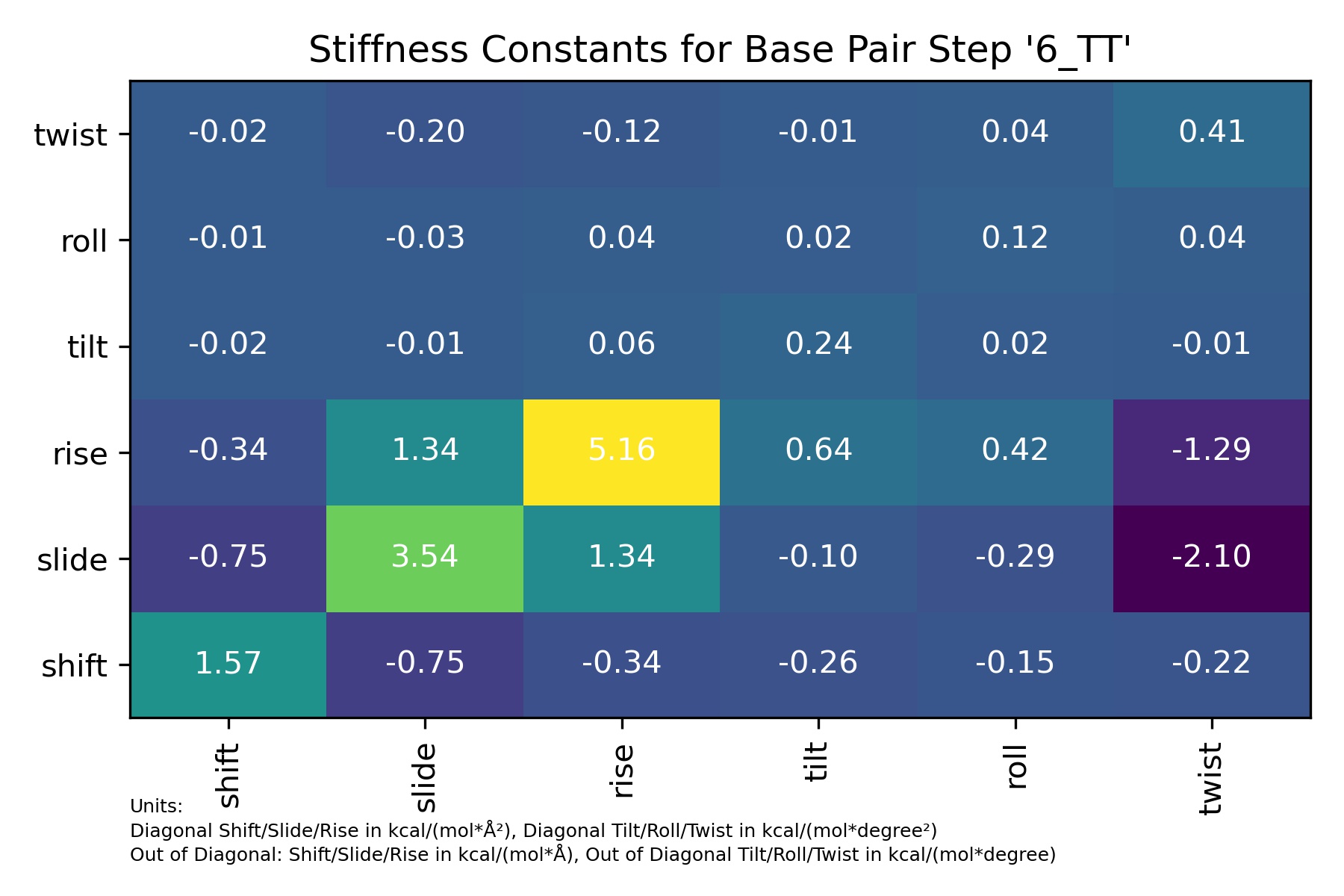
from biobb_dna.stiffness.basepair_stiffness import basepair_stiffness
timeseries_shift = timeseries_dir+"/series_shift_" + timesel.value + ".csv"
timeseries_slide = timeseries_dir+"/series_slide_" + timesel.value + ".csv"
timeseries_rise = timeseries_dir+"/series_rise_" + timesel.value + ".csv"
timeseries_tilt = timeseries_dir+"/series_tilt_" + timesel.value + ".csv"
timeseries_roll = timeseries_dir+"/series_roll_" + timesel.value + ".csv"
timeseries_twist = timeseries_dir+"/series_twist_" + timesel.value + ".csv"
output_stiffness_bps_csv_path = "stiffness_bps.csv"
output_stiffness_bps_jpg_path = "stiffness_bps.jpg"
basepair_stiffness(
input_filename_shift=timeseries_shift,
input_filename_slide=timeseries_slide,
input_filename_rise=timeseries_rise,
input_filename_tilt=timeseries_tilt,
input_filename_roll=timeseries_roll,
input_filename_twist=timeseries_twist,
output_csv_path=output_stiffness_bps_csv_path,
output_jpg_path=output_stiffness_bps_jpg_path)
df = pd.read_csv(output_stiffness_bps_csv_path)
df
| 6_TT | shift | slide | rise | tilt | roll | twist | |
|---|---|---|---|---|---|---|---|
| 0 | shift | 1.565629 | -0.750135 | -0.335147 | -0.263984 | -0.153248 | -0.220935 |
| 1 | slide | -0.750135 | 3.535266 | 1.335823 | -0.095199 | -0.287142 | -2.104566 |
| 2 | rise | -0.335147 | 1.335823 | 5.158742 | 0.637038 | 0.415877 | -1.293672 |
| 3 | tilt | -0.024904 | -0.008981 | 0.060098 | 0.235447 | 0.015406 | -0.008404 |
| 4 | roll | -0.014457 | -0.027089 | 0.039234 | 0.015406 | 0.119149 | 0.038313 |
| 5 | twist | -0.020843 | -0.198544 | -0.122045 | -0.008404 | 0.038313 | 0.412179 |
Image(filename=output_stiffness_bps_jpg_path,width = 600)
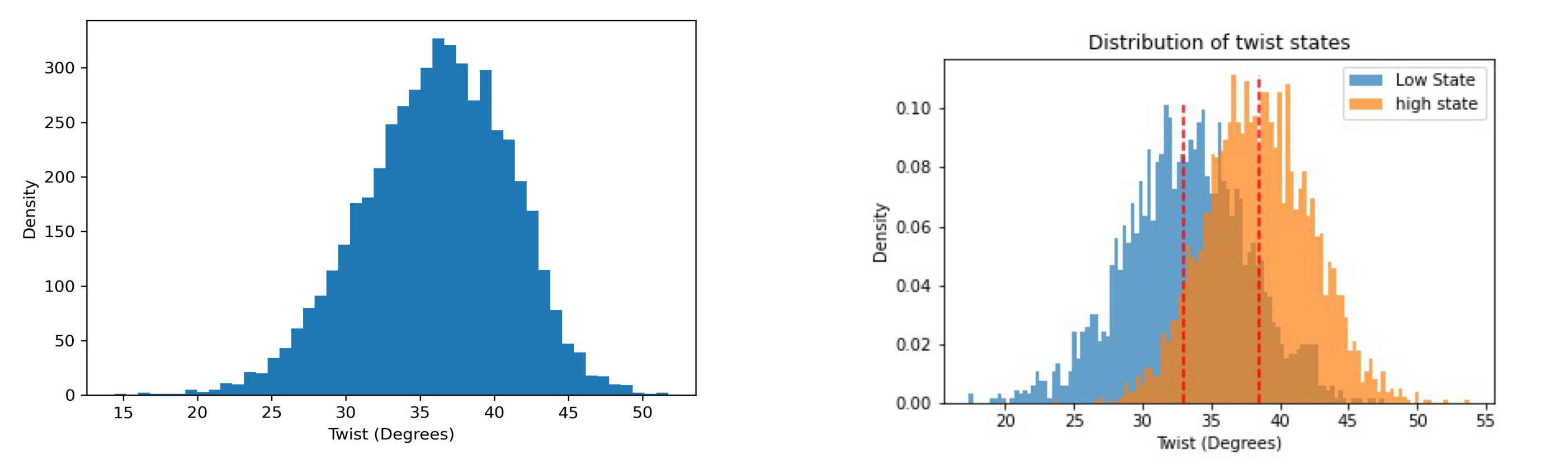
Bimodality
Base-pair steps helical parameters usually follow a normal (Gaussian-like) distribution. However, recent studies observed bimodal distributions in some base-pair steps for twist and slide, highlighting potential caveats on the harmonic approximation implicit in elastic analysis and mesoscopic models of DNA flexibility.

μABC: a systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA
Marco Pasi, John H Maddocks, David Beveridge, Thomas C Bishop, David A Case, Thomas Cheatham 3rd, Pablo D Dans, B Jayaram, Filip Lankas, Charles Laughton, Jonathan Mitchell, Roman Osman, Modesto Orozco, Alberto Pérez, Daiva Petkevičiūtė, Nada Spackova, Jiri Sponer, Krystyna Zakrzewska, Richard Lavery
Nucleic Acids Research 2014, Volume 42, Issue 19, Pages 12272-12283
https://doi.org/10.1093/nar/gku855
Exploring polymorphisms in B-DNA helical conformations
Pablo D Dans, Alberto Pérez, Ignacio Faustino, Richard Lavery, Modesto Orozco
Nucleic Acids Research 2012, Volume 40, Issue 21, Pages 10668-10678
https://doi.org/10.1093/nar/gks884
A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA
Lavery R, Zakrzewska K, Beveridge D, Bishop TC, Case DA, Cheatham T, III, Dixit S, Jayaram B, Lankas F, Laughton C, John H Maddocks, Alexis Michon, Roman Osman, Modesto Orozco, Alberto Perez, Tanya Singh, Nada Spackova, Jiri Sponer
Nucleic Acids Research 2010, Volume 38, Pages 299–313
https://doi.org/10.1093/nar/gkp834
Building Block used:
dna_bimodality from biobb_dna.dna.bimodality
from biobb_dna.dna.dna_bimodality import dna_bimodality
helpar = "twist"
input_csv = timeseries_dir+"/series_"+helpar+"_"+timesel.value+'.csv'
#input_csv = "/Users/hospital/biobb_tutorials/biobb_dna/timeseries"+"/series_"+timesel.value+'.csv' # <-- TO BE REPLACED BY PREVIOUS LINE
output_bimodality_csv = helpar+'.bimodality.csv'
output_bimodality_jpg = helpar+'.bimodality.jpg'
prop = {
'max_iter': 500
}
dna_bimodality(
input_csv_file=input_csv,
output_csv_path=output_bimodality_csv,
output_jpg_path=output_bimodality_jpg,
properties=prop)
file_hist = timeseries_dir + "/hist_" + helpar + "_" + timesel.value + ".jpg"
file_bi = helpar + ".bimodality.jpg"
images = []
images.append(file_hist)
images.append(file_bi)
f, axarr = plt.subplots(1, 2, figsize=(50, 15))
for i, image in enumerate(images):
img = mpimg.imread(image)
axarr[i].imshow(img, aspect='auto')
axarr[i].axis('off')
plt.show()
Correlations
Sequence-dependent correlation movements have been identified in DNA conformational analysis at the base pair and base pair-step level. Trinucleotides were found to show moderate to high correlations in some intra base pair helical parameter (e.g. shear-opening, shear-stretch, stagger-buckle). Similarly, some tetranucleotides are showing strong correlations in their inter base pair helical parameters (e.g. shift-tilt, slide-twist, rise-tilt, shift-slide, and shift-twist in RR steps), with negative correlations in the shift-slide and roll-twist cases. Correlations are also observed in the combination of inter- and intra-helical parameters (e.g. shift-opening, rise-buckle, stagger-tilt). Correlations analysis can be also extended to neighboring steps (e.g. twist in the central YR step of XYRR tetranucleotides with slide in the adjacent RR step).

The static and dynamic structural heterogeneities of B-DNA: extending Calladine–Dickerson rules
Pablo D Dans, Alexandra Balaceanu, Marco Pasi, Alessandro S Patelli, Daiva Petkevičiūtė, Jürgen Walther, Adam Hospital, Genís Bayarri, Richard Lavery, John H Maddocks, Modesto Orozco
Nucleic Acids Research 2019, Volume 47, Issue 21, Pages 11090-11102
https://doi.org/10.1093/nar/gkz905
Building Blocks used:
intraseqcorr from biobb_dna.intrabp_correlations.intraseqcorr
interseqcorr from biobb_dna.interbp_correlations.interseqcorr
intrahpcorr from biobb_dna.intrabp_correlations.intrahpcorr
interhpcorr from biobb_dna.interbp_correlations.interhpcorr
intrabpcorr from biobb_dna.intrabp_correlations.intrabpcorr
interbpcorr from biobb_dna.interbp_correlations.interbpcorr
Sequence Correlations: Intra-base pairs
from biobb_dna.intrabp_correlations.intraseqcorr import intraseqcorr
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_intrabp_correlation_csv_path = helpar+'.intrabp_correlation.csv'
output_intrabp_correlation_jpg_path = helpar+'.intrabp_correlation.jpg'
prop={
'sequence' : seq,
# 'helpar_name' : 'Rise'
}
intraseqcorr(
input_ser_path=input_file_path,
output_csv_path=output_intrabp_correlation_csv_path,
output_jpg_path=output_intrabp_correlation_jpg_path,
properties=prop)
df = pd.read_csv(output_intrabp_correlation_csv_path)
df
| Unnamed: 0 | G | C | G_dup | A | A_dup | T | T_dup | C_dup | G_dup_dup | C_dup_dup | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | G | 1.000000 | -0.358404 | 0.068163 | 0.009405 | 0.001087 | 0.019698 | -0.015957 | 0.007849 | 0.031577 | 0.010621 |
| 1 | C | -0.358404 | 1.000000 | -0.448247 | -0.007771 | 0.026130 | -0.000455 | -0.005333 | -0.013427 | 0.001874 | 0.025165 |
| 2 | G_dup | 0.068163 | -0.448247 | 1.000000 | -0.335686 | -0.048327 | -0.029954 | 0.018416 | 0.013668 | -0.026111 | 0.016239 |
| 3 | A | 0.009405 | -0.007771 | -0.335686 | 1.000000 | -0.293111 | 0.070316 | -0.003760 | 0.006406 | -0.000036 | 0.007260 |
| 4 | A_dup | 0.001087 | 0.026130 | -0.048327 | -0.293111 | 1.000000 | -0.273114 | -0.056474 | 0.001153 | -0.002821 | -0.016981 |
| 5 | T | 0.019698 | -0.000455 | -0.029954 | 0.070316 | -0.273114 | 1.000000 | -0.335193 | -0.006889 | -0.001462 | 0.017912 |
| 6 | T_dup | -0.015957 | -0.005333 | 0.018416 | -0.003760 | -0.056474 | -0.335193 | 1.000000 | -0.447663 | 0.059009 | 0.019932 |
| 7 | C_dup | 0.007849 | -0.013427 | 0.013668 | 0.006406 | 0.001153 | -0.006889 | -0.447663 | 1.000000 | -0.372677 | 0.014933 |
| 8 | G_dup_dup | 0.031577 | 0.001874 | -0.026111 | -0.000036 | -0.002821 | -0.001462 | 0.059009 | -0.372677 | 1.000000 | -0.236134 |
| 9 | C_dup_dup | 0.010621 | 0.025165 | 0.016239 | 0.007260 | -0.016981 | 0.017912 | 0.019932 | 0.014933 | -0.236134 | 1.000000 |
Image(filename=output_intrabp_correlation_jpg_path,width = 600)
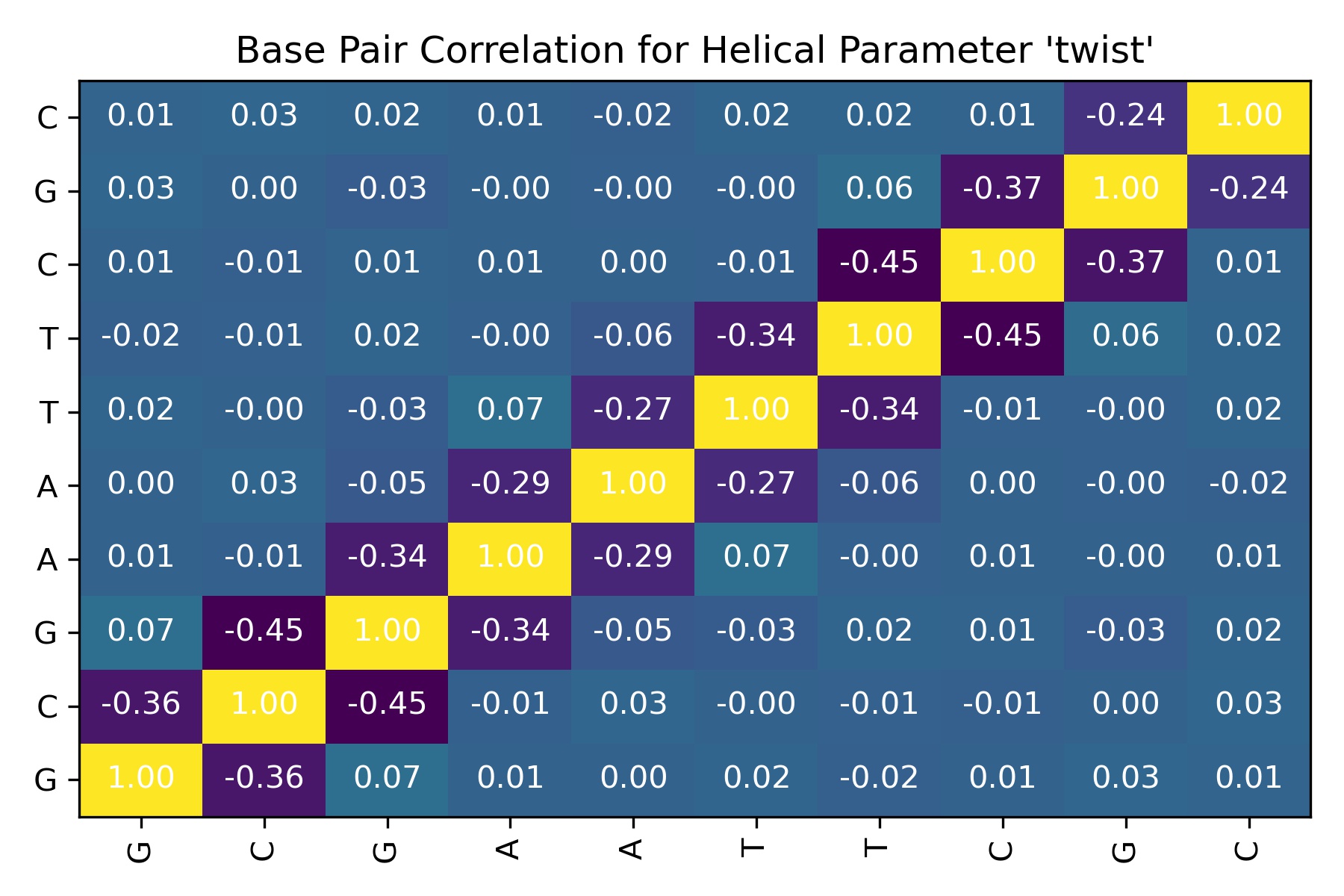
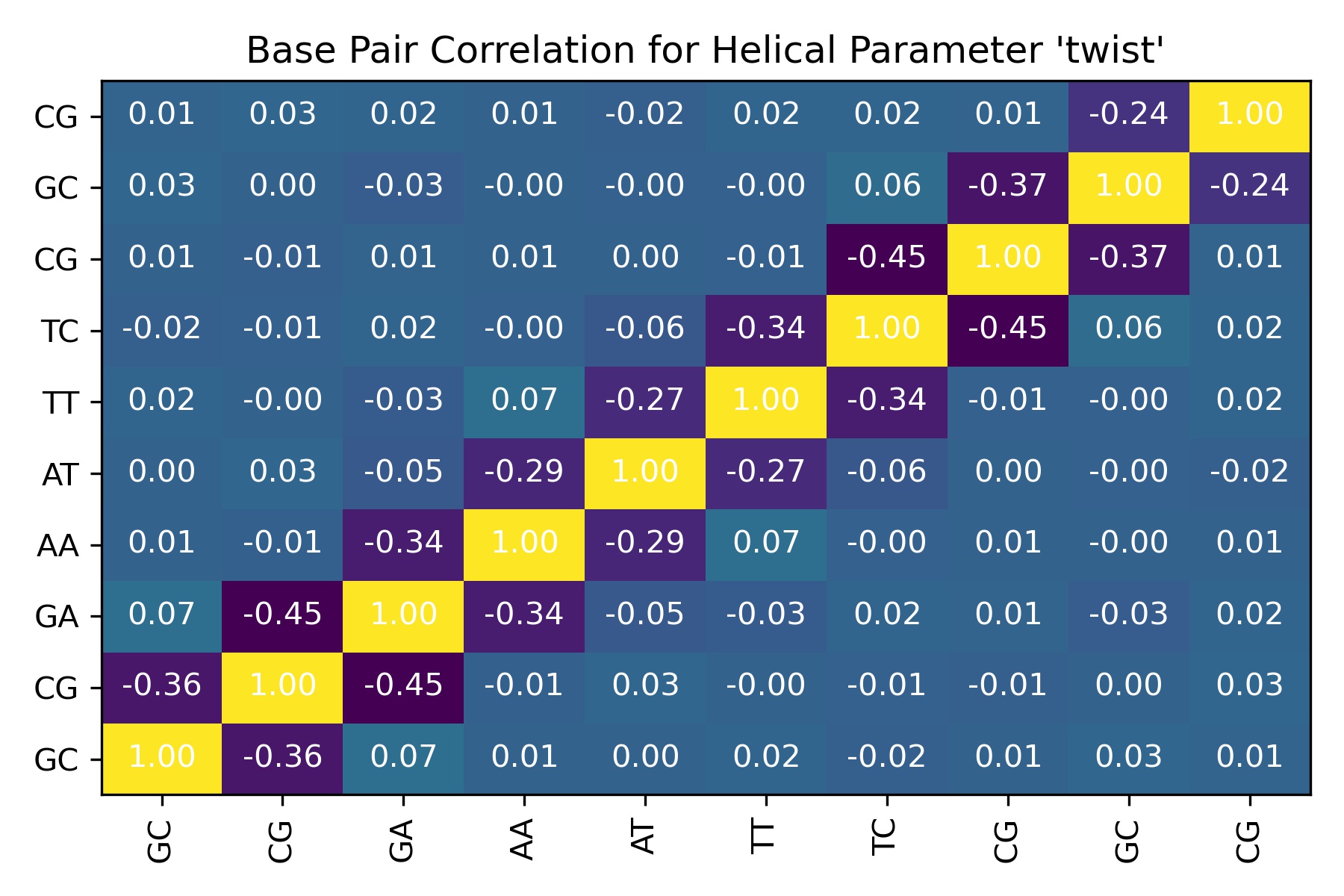
Sequence Correlations: Inter-base pair steps
from biobb_dna.interbp_correlations.interseqcorr import interseqcorr
input_file_path = "canal_out/canal_output" + "_" + helpar + ".ser"
output_interbp_correlation_csv_path = helpar+'.interbp_correlation.csv'
output_interbp_correlation_jpg_path = helpar+'.interbp_correlation.jpg'
prop={
'sequence' : seq,
# 'helpar_name' : 'Rise'
}
interseqcorr(
input_ser_path=input_file_path,
output_csv_path=output_interbp_correlation_csv_path,
output_jpg_path=output_interbp_correlation_jpg_path,
properties=prop)
df = pd.read_csv(output_interbp_correlation_csv_path)
df
| Unnamed: 0 | GC | CG | GA | AA | AT | TT | TC | CG_dup | GC_dup | CG_dup_dup | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | GC | 1.000000 | -0.358404 | 0.068163 | 0.009405 | 0.001087 | 0.019698 | -0.015957 | 0.007849 | 0.031577 | 0.010621 |
| 1 | CG | -0.358404 | 1.000000 | -0.448247 | -0.007771 | 0.026130 | -0.000455 | -0.005333 | -0.013427 | 0.001874 | 0.025165 |
| 2 | GA | 0.068163 | -0.448247 | 1.000000 | -0.335686 | -0.048327 | -0.029954 | 0.018416 | 0.013668 | -0.026111 | 0.016239 |
| 3 | AA | 0.009405 | -0.007771 | -0.335686 | 1.000000 | -0.293111 | 0.070316 | -0.003760 | 0.006406 | -0.000036 | 0.007260 |
| 4 | AT | 0.001087 | 0.026130 | -0.048327 | -0.293111 | 1.000000 | -0.273114 | -0.056474 | 0.001153 | -0.002821 | -0.016981 |
| 5 | TT | 0.019698 | -0.000455 | -0.029954 | 0.070316 | -0.273114 | 1.000000 | -0.335193 | -0.006889 | -0.001462 | 0.017912 |
| 6 | TC | -0.015957 | -0.005333 | 0.018416 | -0.003760 | -0.056474 | -0.335193 | 1.000000 | -0.447663 | 0.059009 | 0.019932 |
| 7 | CG_dup | 0.007849 | -0.013427 | 0.013668 | 0.006406 | 0.001153 | -0.006889 | -0.447663 | 1.000000 | -0.372677 | 0.014933 |
| 8 | GC_dup | 0.031577 | 0.001874 | -0.026111 | -0.000036 | -0.002821 | -0.001462 | 0.059009 | -0.372677 | 1.000000 | -0.236134 |
| 9 | CG_dup_dup | 0.010621 | 0.025165 | 0.016239 | 0.007260 | -0.016981 | 0.017912 | 0.019932 | 0.014933 | -0.236134 | 1.000000 |
Image(filename=output_interbp_correlation_jpg_path,width = 600)
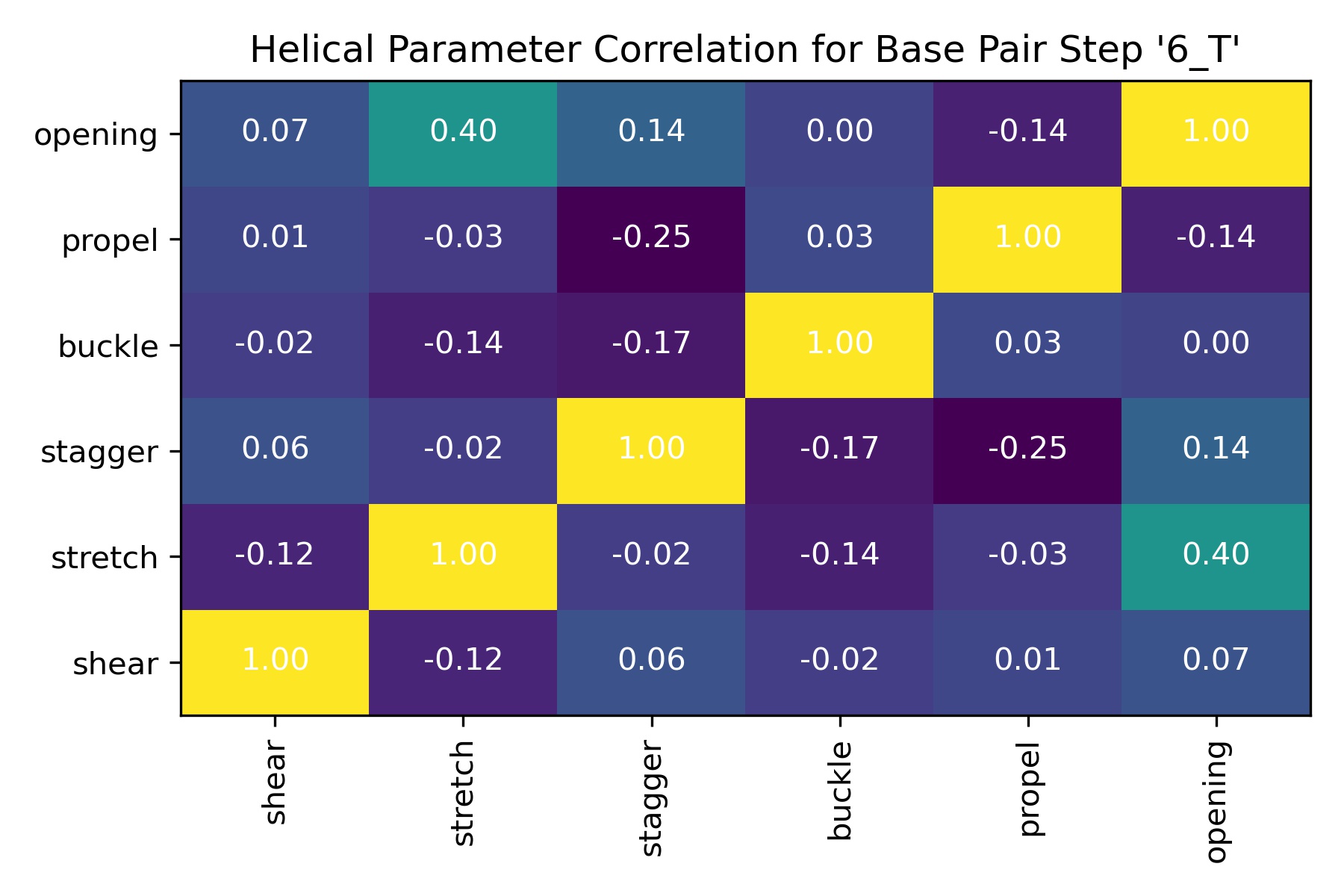
Helical Parameter Correlations: Intra-base pair
from biobb_dna.intrabp_correlations.intrahpcorr import intrahpcorr
timeseries_shear = timeseries_dir+"/series_shear_"+timesel.value[:-1]+".csv"
timeseries_stretch = timeseries_dir+"/series_stretch_"+timesel.value[:-1]+".csv"
timeseries_stagger = timeseries_dir+"/series_stagger_"+timesel.value[:-1]+".csv"
timeseries_buckle = timeseries_dir+"/series_buckle_"+timesel.value[:-1]+".csv"
timeseries_propel = timeseries_dir+"/series_propel_"+timesel.value[:-1]+".csv"
timeseries_opening = timeseries_dir+"/series_opening_"+timesel.value[:-1]+".csv"
output_helpar_bp_correlation_csv_path = "helpar_bp_correlation.csv"
output_helpar_bp_correlation_jpg_path = "helpar_bp_correlation.jpg"
intrahpcorr(
input_filename_shear=timeseries_shear,
input_filename_stretch=timeseries_stretch,
input_filename_stagger=timeseries_stagger,
input_filename_buckle=timeseries_buckle,
input_filename_propel=timeseries_propel,
input_filename_opening=timeseries_opening,
output_csv_path=output_helpar_bp_correlation_csv_path,
output_jpg_path=output_helpar_bp_correlation_jpg_path)
df = pd.read_csv(output_helpar_bp_correlation_csv_path)
df
| Unnamed: 0 | shear | stretch | stagger | buckle | propel | opening | |
|---|---|---|---|---|---|---|---|
| 0 | shear | 1.000000 | -0.122310 | 0.062783 | -0.024045 | 0.012255 | 0.066297 |
| 1 | stretch | -0.122310 | 1.000000 | -0.024948 | -0.141840 | -0.034225 | 0.396172 |
| 2 | stagger | 0.062783 | -0.024948 | 1.000000 | -0.172773 | -0.253086 | 0.138571 |
| 3 | buckle | -0.024045 | -0.141840 | -0.172773 | 1.000000 | 0.029668 | 0.000972 |
| 4 | propel | 0.012255 | -0.034225 | -0.253086 | 0.029668 | 1.000000 | -0.138484 |
| 5 | opening | 0.066297 | 0.396172 | 0.138571 | 0.000972 | -0.138484 | 1.000000 |
Image(filename=output_helpar_bp_correlation_jpg_path,width = 600)
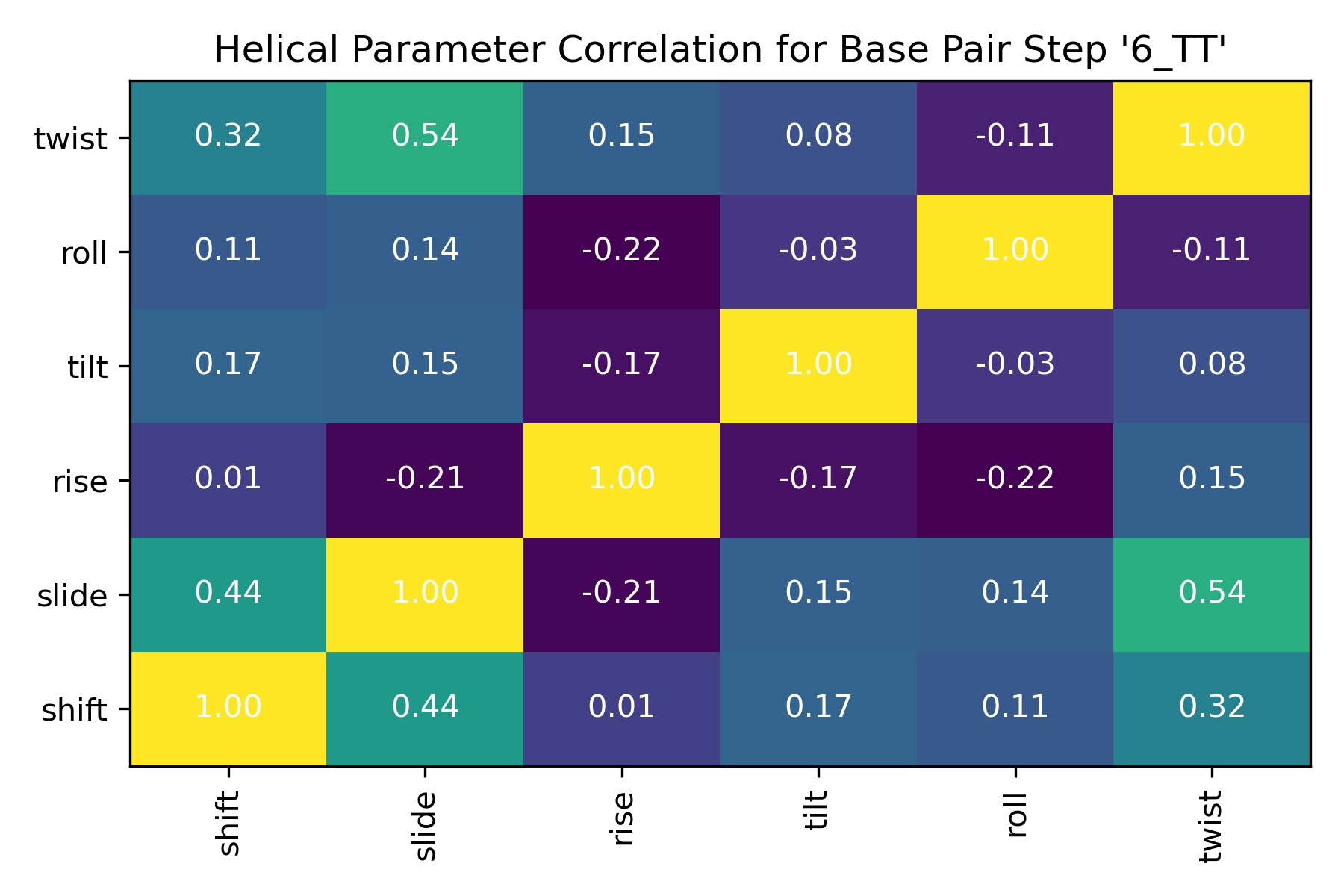
Helical Parameter Correlations: Inter-base pair steps
from biobb_dna.interbp_correlations.interhpcorr import interhpcorr
timeseries_shift = timeseries_dir+"/series_shift_"+timesel.value+".csv"
timeseries_slide = timeseries_dir+"/series_slide_"+timesel.value+".csv"
timeseries_rise = timeseries_dir+"/series_rise_"+timesel.value+".csv"
timeseries_tilt = timeseries_dir+"/series_tilt_"+timesel.value+".csv"
timeseries_roll = timeseries_dir+"/series_roll_"+timesel.value+".csv"
timeseries_twist = timeseries_dir+"/series_twist_"+timesel.value+".csv"
output_helpar_bps_correlation_csv_path = "helpar_bps_correlation.csv"
output_helpar_bps_correlation_jpg_path = "helpar_bps_correlation.jpg"
interhpcorr(
input_filename_shift=timeseries_shift,
input_filename_slide=timeseries_slide,
input_filename_rise=timeseries_rise,
input_filename_tilt=timeseries_tilt,
input_filename_roll=timeseries_roll,
input_filename_twist=timeseries_twist,
output_csv_path=output_helpar_bps_correlation_csv_path,
output_jpg_path=output_helpar_bps_correlation_jpg_path)
df = pd.read_csv(output_helpar_bps_correlation_csv_path)
df
| Unnamed: 0 | shift | slide | rise | tilt | roll | twist | |
|---|---|---|---|---|---|---|---|
| 0 | shift | 1.000000 | 0.439831 | 0.006659 | 0.166307 | 0.113906 | 0.318624 |
| 1 | slide | 0.439831 | 1.000000 | -0.207299 | 0.151236 | 0.141627 | 0.541202 |
| 2 | rise | 0.006659 | -0.207299 | 1.000000 | -0.173336 | -0.224263 | 0.147534 |
| 3 | tilt | 0.166307 | 0.151236 | -0.173336 | 1.000000 | -0.032611 | 0.084610 |
| 4 | roll | 0.113906 | 0.141627 | -0.224263 | -0.032611 | 1.000000 | -0.109835 |
| 5 | twist | 0.318624 | 0.541202 | 0.147534 | 0.084610 | -0.109835 | 1.000000 |
Image(filename=output_helpar_bps_correlation_jpg_path,width = 600)
Neighboring steps Correlations: Intra-base pair
from biobb_dna.intrabp_correlations.intrabpcorr import intrabpcorr
canal_shear = canal_dir+"/canal_output_shear.ser"
canal_stretch = canal_dir+"/canal_output_stretch.ser"
canal_stagger = canal_dir+"/canal_output_stagger.ser"
canal_buckle = canal_dir+"/canal_output_buckle.ser"
canal_propel = canal_dir+"/canal_output_propel.ser"
canal_opening = canal_dir+"/canal_output_opening.ser"
output_bp_correlation_csv_path = "bp_correlation.csv"
output_bp_correlation_jpg_path = "bp_correlation.jpg"
prop = {
'sequence' : seq
}
intrabpcorr(
input_filename_shear=canal_shear,
input_filename_stretch=canal_stretch,
input_filename_stagger=canal_stagger,
input_filename_buckle=canal_buckle,
input_filename_propel=canal_propel,
input_filename_opening=canal_opening,
output_csv_path=output_bp_correlation_csv_path,
output_jpg_path=output_bp_correlation_jpg_path,
properties=prop)
df = pd.read_csv(output_bp_correlation_csv_path)
df
| Unnamed: 0 | shear/shear | shear/stretch | shear/stagger | shear/buckle | shear/propel | shear/opening | stretch/shear | stretch/stretch | stretch/stagger | ... | propel/stagger | propel/buckle | propel/propel | propel/opening | opening/shear | opening/stretch | opening/stagger | opening/buckle | opening/propel | opening/opening | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | GC | -0.005642 | 0.018630 | -0.025087 | -0.010539 | 0.024243 | 0.019345 | -0.011061 | 0.021519 | -0.020824 | ... | -0.024222 | 0.005585 | 0.006706 | -0.021960 | -0.010185 | 0.006839 | -0.011742 | 0.001443 | 0.005266 | 0.019129 |
| 1 | CG | 0.020041 | 0.051434 | 0.001951 | -0.059043 | 0.027216 | 0.045336 | -0.051213 | -0.033895 | 0.009731 | ... | 0.047045 | -0.335039 | -0.122872 | -0.067494 | -0.047289 | -0.037659 | 0.023410 | -0.105277 | -0.082899 | 0.005289 |
| 2 | GA | -0.037801 | 0.027801 | -0.015043 | -0.105601 | 0.073992 | 0.025790 | -0.010322 | 0.043308 | 0.011708 | ... | 0.073619 | -0.176741 | -0.051962 | 0.003138 | 0.029520 | 0.005160 | 0.051622 | 0.007026 | -0.017732 | 0.021906 |
| 3 | AA | 0.023788 | -0.000117 | -0.004595 | -0.027747 | -0.031901 | -0.041804 | 0.023817 | -0.006556 | -0.005822 | ... | -0.061579 | -0.147203 | -0.004269 | -0.050649 | -0.039697 | 0.035455 | -0.018540 | -0.093678 | -0.048917 | 0.042879 |
| 4 | AT | 0.032575 | 0.003270 | -0.028156 | -0.070385 | -0.066959 | 0.021602 | 0.014530 | 0.014772 | -0.047132 | ... | -0.113304 | -0.173226 | 0.107649 | 0.019869 | -0.036929 | -0.020634 | -0.030510 | 0.021788 | 0.036952 | 0.119437 |
| 5 | TT | -0.010285 | -0.009504 | 0.035414 | 0.004137 | 0.003110 | 0.052842 | -0.020711 | 0.005863 | -0.007229 | ... | -0.091332 | -0.132041 | 0.183310 | 0.023056 | -0.051025 | 0.023035 | -0.017409 | 0.003068 | 0.010977 | 0.191239 |
| 6 | TC | 0.002390 | 0.003433 | -0.024093 | 0.014480 | -0.012981 | 0.059420 | 0.030606 | 0.011428 | 0.005235 | ... | -0.019650 | -0.152708 | 0.080905 | 0.021846 | -0.012044 | 0.067861 | -0.083172 | 0.048029 | 0.028434 | 0.151051 |
| 7 | CG | -0.027842 | 0.002501 | 0.014905 | 0.010619 | 0.018963 | 0.030263 | -0.020690 | 0.016758 | 0.033150 | ... | 0.063238 | -0.222382 | 0.012536 | -0.021479 | -0.042303 | 0.020077 | 0.014984 | -0.018558 | -0.076957 | 0.006822 |
| 8 | GC | -0.030918 | 0.025377 | 0.047997 | -0.149693 | 0.032799 | 0.004369 | -0.029652 | 0.018781 | 0.037963 | ... | 0.063868 | -0.188492 | -0.044925 | -0.027264 | -0.025809 | 0.027768 | 0.030092 | -0.062296 | 0.025229 | 0.019334 |
| 9 | CG | -0.001727 | 0.039837 | 0.013472 | -0.031293 | -0.014562 | 0.043469 | -0.018693 | -0.008573 | 0.026310 | ... | 0.044891 | -0.303367 | -0.113362 | -0.080200 | -0.018591 | -0.029267 | 0.032287 | -0.095808 | -0.074735 | -0.005167 |
10 rows × 37 columns
Image(filename=output_bp_correlation_jpg_path,width = 800)
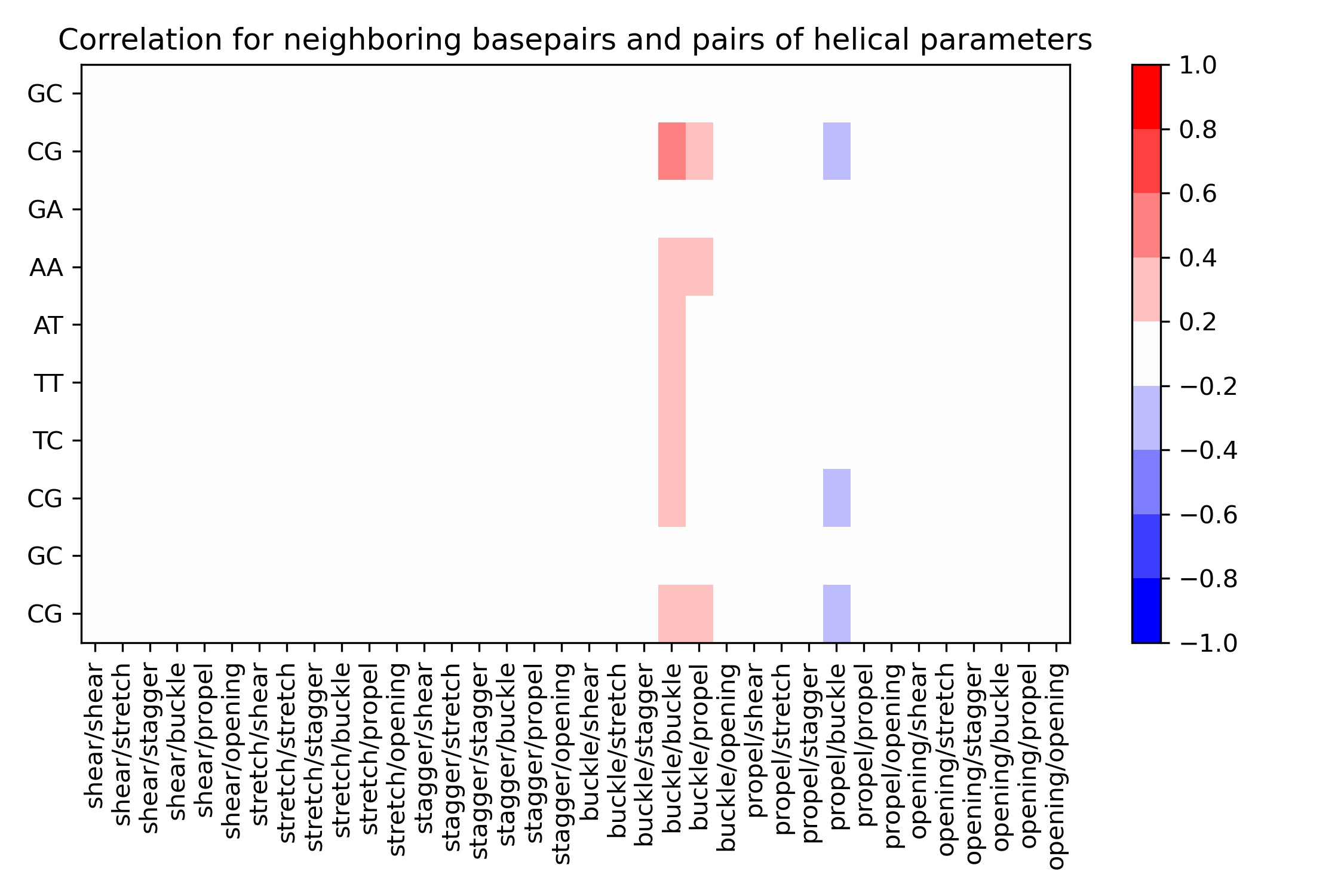
Neighboring steps Correlations: Inter-base pair steps
from biobb_dna.interbp_correlations.interbpcorr import interbpcorr
canal_shift = canal_dir+"/canal_output_shift.ser"
canal_slide = canal_dir+"/canal_output_slide.ser"
canal_rise = canal_dir+"/canal_output_rise.ser"
canal_tilt = canal_dir+"/canal_output_tilt.ser"
canal_roll = canal_dir+"/canal_output_roll.ser"
canal_twist = canal_dir+"/canal_output_twist.ser"
output_bps_correlation_csv_path = "bps_correlation.csv"
output_bps_correlation_jpg_path = "bps_correlation.jpg"
prop = {
'sequence' : seq
}
interbpcorr(
input_filename_shift=canal_shift,
input_filename_slide=canal_slide,
input_filename_rise=canal_rise,
input_filename_tilt=canal_tilt,
input_filename_roll=canal_roll,
input_filename_twist=canal_twist,
output_csv_path=output_bps_correlation_csv_path,
output_jpg_path=output_bps_correlation_jpg_path,
properties=prop)
df = pd.read_csv(output_bps_correlation_csv_path)
df
| Unnamed: 0 | shift/shift | shift/slide | shift/rise | shift/tilt | shift/roll | shift/twist | slide/shift | slide/slide | slide/rise | ... | roll/rise | roll/tilt | roll/roll | roll/twist | twist/shift | twist/slide | twist/rise | twist/tilt | twist/roll | twist/twist | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | GCG | -0.034515 | -0.013978 | 0.028504 | -0.016774 | 0.007526 | -0.019706 | -0.005945 | 0.003258 | 0.016998 | ... | -0.024289 | 0.034074 | -0.014922 | 0.002918 | 0.024087 | 0.011902 | -0.018600 | -0.000658 | 0.004749 | 0.031633 |
| 1 | CGA | -0.620729 | -0.213534 | 0.153552 | -0.379811 | -0.069482 | 0.090324 | -0.185796 | 0.035204 | -0.072609 | ... | 0.185182 | 0.239957 | -0.327237 | 0.146238 | -0.301986 | -0.066687 | -0.230273 | -0.149935 | 0.234940 | -0.358276 |
| 2 | GAA | -0.556258 | -0.055634 | 0.106571 | -0.307558 | -0.124963 | 0.111320 | 0.336513 | 0.207062 | 0.010327 | ... | 0.243220 | 0.222142 | -0.356105 | 0.262547 | -0.112359 | -0.169531 | -0.267369 | -0.017553 | 0.222109 | -0.448559 |
| 3 | AAT | -0.422776 | 0.121578 | 0.172917 | -0.120018 | -0.234489 | 0.333776 | 0.200294 | 0.055308 | -0.062561 | ... | 0.182963 | 0.269344 | -0.261031 | 0.037170 | -0.055827 | -0.035366 | -0.125160 | -0.008524 | 0.080896 | -0.336058 |
| 4 | ATT | -0.135159 | 0.048764 | 0.052558 | 0.152407 | -0.137398 | 0.093877 | -0.058611 | 0.177430 | 0.140074 | ... | 0.189502 | 0.241552 | -0.262516 | 0.016017 | -0.027548 | -0.024130 | -0.059852 | -0.003261 | 0.031997 | -0.293106 |
| 5 | TTC | -0.115228 | 0.074008 | 0.011805 | 0.204128 | -0.216200 | -0.032101 | -0.028032 | 0.184504 | 0.114301 | ... | 0.113645 | 0.247625 | -0.299766 | -0.015864 | -0.148998 | 0.111239 | 0.075143 | 0.053240 | -0.015885 | -0.273020 |
| 6 | TCG | -0.428290 | -0.230813 | 0.116441 | 0.057122 | -0.208447 | -0.118440 | -0.121029 | 0.055468 | 0.090725 | ... | 0.093449 | 0.287541 | -0.273905 | 0.101453 | -0.316904 | -0.253028 | -0.076103 | -0.114819 | 0.066582 | -0.335519 |
| 7 | CGC | -0.577276 | -0.389311 | 0.286179 | -0.324285 | -0.179625 | 0.071846 | 0.054698 | 0.233093 | -0.148480 | ... | 0.140274 | 0.210203 | -0.362062 | 0.244422 | -0.150061 | -0.097582 | -0.265051 | -0.021533 | 0.240612 | -0.447802 |
| 8 | GCG | -0.626682 | 0.229957 | 0.290226 | -0.338901 | -0.240202 | 0.328321 | 0.213949 | 0.040634 | 0.007077 | ... | 0.287660 | 0.196857 | -0.327687 | 0.263437 | -0.127565 | -0.149207 | -0.200700 | -0.092674 | 0.171328 | -0.372676 |
9 rows × 37 columns
Image(filename=output_bps_correlation_jpg_path,width = 800)
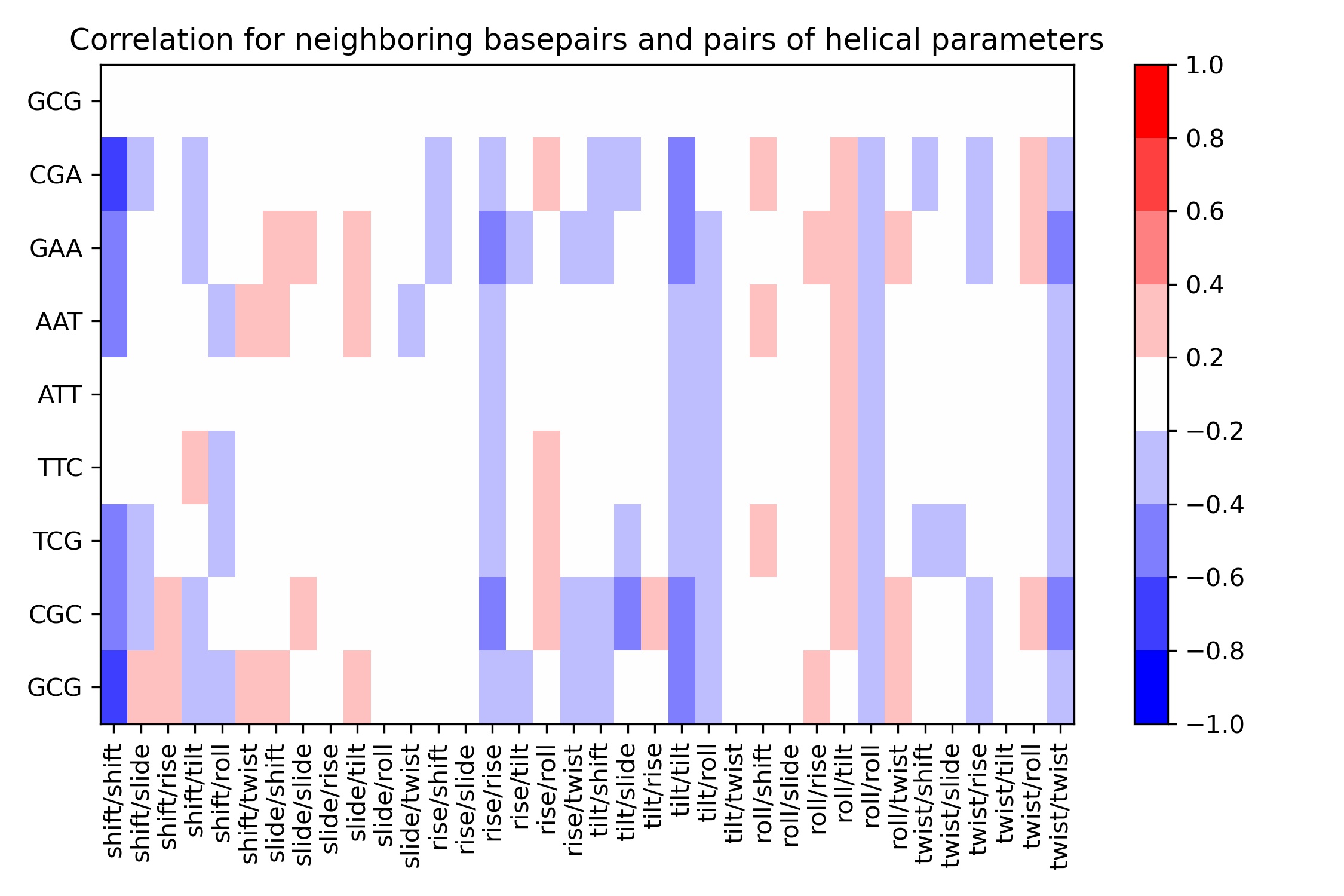
Questions & Comments
Questions, issues, suggestions and comments are really welcome!
GitHub issues:
BioExcel forum: